
| http://www.ccl.net/cca/documents/dyoung/topics-framed/compchem.shtml |

|
CCL compchem.html |
Introduction to Computational Chemistry
David Young
Table of Contents
IntroductionRecent years have seen an increase in the number of people doing theoretical chemistry. Many of these newcomers are part time theoreticians, who work on other aspects of chemistry as well. This increase has been facilitated by the development of computer software which is increasingly easy to use. It is now easy enough to do computational chemistry that you do not have to know what you are doing to do a computation. As a result, many people don't understand even the most basic description of how the calculation is done and are therefore sucessufully doing a lot of work which is, frankly, garbage.Many universities are now offering classes, which are an overview of various aspects of computational chemistry. Since we have had many people wanting to start doing computations before they have had even an introductory course, this document has been written as step one in understanding what computational chemistry is about. Note that this is not intended to teach the fundamentals of chemistry, quantum mechanics or mathematics, only most basic description of how chemical computations are done. The term theoretical chemistry may be defined as the mathematical description of chemistry. The term computational chemistry is usually used when a mathematical method is sufficiently well developed that it can be automated for implementation on a computer. Note that the words exact and perfect do not appear in these definitions. Very few aspects of chemistry can be computed exactly, but almost every aspect of chemistry has been described in a qualitative or approximate quantitative computational scheme. The biggest mistake that a computational chemists can make is to assume that any computed number is exact. However, just as not all spectra are perfectly resolved, often a qualitative or approximate computation can give useful insight into chemistry if you understand what it tells you and what it doesn't. Although most chemists avoid the true paper & pencil type of theoretical chemistry, keep in mind that this is what many Nobel prizes have been awarded for.
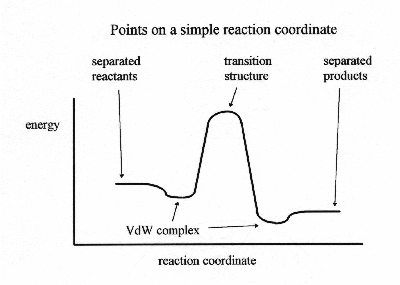
Ab InitioThe term "Ab Initio" is latin for "from the beginning". This name is given to computations which are derived directly from theoretical principles, with no inclusion of experimental data. Most of the time this is referring to an approximate quantum mechanical calculation. The approximations made are usually mathematical approximations, such as using a simpler functional form for a function or getting an approximate solution to a differential equation.The most common type of ab initio calculation is called a Hartree Fock calculation (abbreviated HF), in which the primary approximation is called the central field approximation. This means that the Coulombic electron-electron repulsion is not specifically taken into account. However, it's net effect is included in the calculation. This is a variational calculation, meaning that the approximate energies calculated are all equal to or greater than the exact energy. The energies calculated are usually in units called Hartrees (1 H = 27.2114 eV). Because of the central field approximation, the energies from HF calculations are always greater than the exact energy and tend to a limiting value called the Hartree Fock limit. The second approximation in HF calculations is that the wave function must be described by some functional form, which is only known exactly for a few one electron systems. The functions used most often are linear combinations of Slater type orbitals exp(-ax) or Gaussian type orbitals exp(-ax^2), abbreviated STO and GTO. The wave function is formed from linear combinations of atomic orbitals or more often from linear combinations of basis functions. Because of this approximation, most HF calculations give a computed energy greater than the Hartree Fock limit. The exact set of basis functions used is often specified by an abbreviation, such as STO-3G or 6-311++g**. A number of types of calculations begin with a HF calculation then correct for the explicit electron-electron repulsion, referred to as correlation. Some of these methods are Mohlar-Plesset perturbation theory (MPn, where n is the order of correction), the Generalized Valence Bond (GVB) method, Multi-Configurations Self Consistent Field (MCSCF), Configuration Interaction (CI) and Coupled Cluster theory (CC). As a group, these methods are referred to as correlated calculations. A method, which avoids making the HF mistakes in the first place is called Quantum Monte Carlo (QMC). There are several flavors of QMC .. variational, diffusion and Green's functions. These methods work with an explicitly correlated wave function and evaluate integrals numerically using a Monte Carlo integration. These calculations can be very time consuming, but they are probably the most accurate methods known today. An alternative ab initio method is Density Functional Theory (DFT), in which the total energy is expressed in terms of the total electron density, rather than the wavefunction. In this type of calculation, there is an approximate Hamiltonian and an approximate expression for the total electron density. The good side of ab initio methods is that they eventually converge to the exact solution, once all of the approximations are made sufficiently small in magnitude. However, this convergence is not montonic. Sometimes, the smallest calculation gives the best result for a given property. The bad side of ab initio methods is that they are expensive. These methods often take enormous amounts of computer cpu time, memory and disk space. The HF method scales as N4, where N is the number of basis functions, so a calculation twice as big takes 16 times as long to complete. Correlated calculations often scale much worse than this. In practice, extremely accurate solutions are only obtainable when the molecule contains half a dozen electrons or less. In general, ab initio calculations give very good qualitative results and can give increasingly accurate quantitative results as the molecules in question become smaller.
SemiempiricalSemiempirical calculations are set up with the same general structure as a HF calculation. Within this framework, certain pieces of information, such as two electron integrals, are approximated or completely omitted. In order to correct for the errors introduced by omitting part of the calculation, the method is parameterized, by curve fitting in a few parameters or numbers, in order to give the best possible agreement with experimental data.The good side of semiempirical calculations is that they are much faster than the ab initio calculations. The bad side of semiempirical calculations is that the results can be eratic. If the molecule being computed is similar to molecules in the data base used to parameterize the method, then the results may be very good. If the molecule being computed is significantly different from anything in the parameterization set, the answers may be very poor. Semiempirical calculations have been very successful in the description of organic chemistry, where there are only a few elements used extensively and the molecules are of moderate size. However, semiempirical methods have been devised specifically for the description of inorganic chemistry as well.
Modeling the solid stateThe electronic structure of an infinite crystal is defined by a band structure plot, which gives energies of electron orbitals for each point in k-space, called the Brillouin zone. Since ab initio and semiempirical calculations yield orbital energies, they can be applied to band structure calculations. However, if it is time consuming to calculate the energy for a molecule, it is even more time consuming to calculate energies for a list of points in the Brillouin zone.Band structure calculations have been done for very complicated systems, however the software is not yet automated enough or sufficiently fast that anyone does band structures casually. If you want to do band structure calculations, you had better expect to put a lot of time into your efforts.
Molecular MechanicsIf a molecule is too big to effectively use a semiempirical treatment, it is still possible to model it's behavior by avoiding quantum mechanics totally. The methods referred to as Molecular Mechanics set up a simple algebraic expression for the total energy of a compound, with no necessity to compute a wave function or total electron density. The energy expression consists of simple classical equations, such as the harmonic oscillator equation in order to describe the energy associated with bond stretching, bending, rotation and intermolecular forces, such as van der waals interactions and hydrogen bonding. All of the constants in these equations must be obtained from experimental data or an ab initio calculation.In a molecular mechanics method, the data base of compounds used to parameterize the method (a set of parameters and functions is called a force field) is crucial to it's success. Where as a semiempirical method may be parameterized against a set of organic molecules, a molecular mechanics method may be parameterized against a specific class of molecules, such as proteins. Such a force field would only be expected to have any relevance to describing other proteins. The good side of molecular mechanics is that it allows the modeling of enormous molecules, such as proteins and segments of DNA, making it the primary tool of computational biochemists. The bad side of molecular mechanics is that there are many chemical properties that are not even defined within the method, such as electronic excited states. In order to work with extremely large and complicated systems, often molecular mechanics software packages have the most powerful and easiest to use graphical interfaces. Because of this, mechanics is sometimes used because it is easy, but not necessarily a good way to describe a system.
Molecular DynamicsMolecular dynamics consists of examining the time dependent behavior of a molecule, such as vibrational motion or Brownian motion. This is most often done within a classical mechanical description similar to a molecular mechanics calculation.The application of molecular dynamics to solvent/solute systems allows the computation of properties such as diffusion coeficients or radial distribution functions for use in statistical mechanical treatments. Usually the scheme of a solvent/solute calculation is that a number of molecules (perhaps 1000) are given some initial position and velocity. New positions are calculated a small time later based on this movement and this process is itterated for thousands of steps in order to bring the system to equilibrium and give a good statistical description of the radial distribution function. In order to analyze the vibrations of a single molecule, many dynamics steps are done, then the data is Fourier transformed into the frequency domain. A given peak can be chosen and transformed back to the time domain, in order to see what the motion at that frequency looks like.
Statistical MechanicsStatistical mechanics is the mathematical means to extrapolate thermodynamic properties of bulk materials from a molecular description of the material. Much of statistical mechanics is still at the paper and pencil stage of theory, since the quantum mechanicians can't solve the Schrödinger equation exactly yet, the statistical mechanicians don't really have even a good starting point for a truly rigorous treatment. Statistical mechanics computations are often tacked onto the end of ab inito calculations for gas phase properties. For condensed phase properties, often molecular dynamics calculations are necessary in order to do a computational experiment.
ThermodynamicsThermodynamics is one of the most well developed mathematical chemical descriptions. Very often any thermodynamic treatment is left for trivial pen and paper work since many aspects of chemistry are so accurately described with very simple mathematical expressions.
Structure-Property RelationshipsStructure-property relationships are qualitative or quantitative empirically defined relationships between molecular structure and observed properties. In some cases this may seem to duplicate statistical mechanical results, however structure-property relationships need not be based on any rigorous theoretical principles.The simplest case of structure-property relationships are qualitative thumb rules. For example, an experienced polymer chemist may be able to predict whether a polymer will be soft or brittle based on the geometry and bonding of the monomers. When structure-property relationships are mentioned in current literature, it usually implies a quantitative mathematical relationship. These relationships are most often derived by using curve fitting software to find the linear combination of molecular properties, which best reproduces the desired property. The molecular properties are usually obtained from molecular modeling computations. Other molecular descriptors such as molecular weight or topological descriptions are also used. When the property being described is a physical property, such as the boiling point, this is refered to as a Quantitative Structure-Property Relationship (QSPR). When the property being described is a type of biological activity (such as drug activity), this is refered to as a Quantitative Structure-Activity Relationship (QSAR).
Symbolic CalculationsSymbolic calculations are performed when the system is just too large for an atom-by-atom description to be viable at any level of approximation. An example might be the description of a membrane by describing the individual lipids as some representative polygon with some expression for the energy of interaction. This sort of treatment is used for computational biochemistry and even microbiology.
Artifical IntelligenceTechniques invented by computer scientists interested in artificial intelligence have been applied mostly to drug design in recent years. These methods also go by the names De Novo or rational drug design. The general scenario is that some functional site has been identified and it is desired to come up with a structure for a molecule that will interact with that site in order to hinder it's functionality. Rather than have a chemist try hundreds or thousands of possibilities with a molecular mechanics program, the molecular mechanics is built into an artificial intelligence program, which tries enormous numbers of "reasonable" possibilities in an automated fasion. The number of techniques for describing the "intelligent" part of this operation are so diverse that it is impossible to make any generalization about how this is implemented in the program.
How to do a computational research projectWhen using computational chemistry to answer a chemical question, the obvious problem is that you need to know how to use the software. The problem that is missed is that you need to know how good the answer is going to be. Here is a check list to follow.What do you want to know? How accurately? Why? If you can't answer these questions, then you don't even have a research project yet. How accurate do you predict the answer will be? In analytical chemistry, you do a number of identical measurements then work out the error from a standard deviation. With computational experiments, doing the same thing should always give exactly the same result. The way that you estimate your error is to compare a number of similar computations to the experimental answers. There are articles and compilations of these studies. If none exist, you will have to guess which method should be reasonable, based on it's assumptions then do a study yourself, before you can apply it to you unknown and have any idea how good the calculation is. When someone just tells you off the top of their head what method to use, they either have a fair amount of this type of information memorized, or they don't know what they are talking about. Beware of someone who tells you a given program is good just because it is the only one they know how to use, rather than the basing their answer on the quality of the results. How long do you expect it to take? If the world were perfect, you would tell your PC (voice input of course) to give you the exact solution to the Schrödinger equation and go on with your life. However, often ab initio calculations would be so time consuming that it would take a decade to do a single calculation, if you even had a machine with enough memory and disk space. However, a number of methods exist because each is best for some situation. The trick is to determine which one is best for your project. Again, the answer is to look into the literature and see how long each takes. If the only thing you know is how a calculation scales, do the simplest possible calculation then use the scaling equation to estimate how long it will take to do the sort of calculation that you have predicted will give the desired accuracy. What approximations are being made? Which are significant? This is how you avoid looking like a complete fool, when you successfully perform a calculation that is complete garbage. An example would be trying to find out about vibrational motions that are very anharmonic, when the calculation uses a harmonic oscillator approximation. Once you have finally answered all of these questions, you are ready to actually do a calculation. Now you must determine what software is available, what it costs and how to use it. Note that two programs of the same type (i.e. ab initio) may calculate different properties, so you have to make sure the program does exactly what you want. When you are learning how to use a program, you may try to do dozens of calculations that will fail because you constructed the input incorrectly. Do not use your project molecule to do this. Make all your mistakes with something really easy, like a water molecule. That way you don't waste enormous amounts of time.
VisualizationData visualization is the process of displaying information in any sort of pictorial or graphical representation. A number of computer programs are now available to apply a colorization scheme to data or work with three dimensional representations.
Further informationFor an introductory level overview of computational chemistry seeG. H. Grant, W. G. Richards "Computational Chemistry" Oxford (1995)
A more detailed description of common computational chemistry techniques
is contained in
There are many books on the principles of quantum mechanics and every
physical chemistry text has an introductory treatment. The
work which I am listing here is a two volume set with each chapter
broken into a basic and advanced sections making it excellent for
both intermediate and advanced users.
For an introduction to quantum chemistry see
A graduate level text on quantum chemistry is
An advanced undergraduate or graduate text on quantum chemistry is
For quantum Monte Carlo methods, order the following book using
ISBN 981-02-0322-5 because the title is listed incorrectly in
'Books in Print'.
A good review article on density functional theory is
For density functional theory see
For a basic understanding of solid state modeling see
For a graduate level description of statistical mechanics see
Any physical chemistry text will have a description of thermodynamics
but I will recommend
Another nice introduction to computational chemistry is
There is a comprehensive listing of all available molecular modeling
software and structural databanks, free or not, in appendix 2 of
There is a write up on computer aided drug design at
Mathematical challenges from theoretical/computational chemistry
An online text on molecular modeling using molecular mechanics
A Computational Chemistry Primer
An online text on computational chemistry
Another online text on quantum chemistry
An online introduction to quantum mechanics is at Citation: This article was originally published on the web. It has now appeared in print in D. Young, Chem. Aust. 11, 5 (1998). An expanded version of this article will be published in "Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems" by David Young, which will be available from John Wiley & Sons in the spring of 2001. |
[ CCL Home Page ]
[ topics ]
[ Raw Version of this page ]
| Modified: Thu Jan 4 02:29:23 2001 GMT |
| Page accessed 11765 times since Sat Jan 6 07:49:01 2001 GMT |
{kind=link}
{kind=link}
{kind=link}
{kind=link}