
| http://www.ccl.net/cca/documents/dyoung/topics-framed/vib.shtml |

|
CCL vib.html |
Computational Modeling of Molecular VibrationsDavid YoungCytoclonal Pharmaceutics Inc.
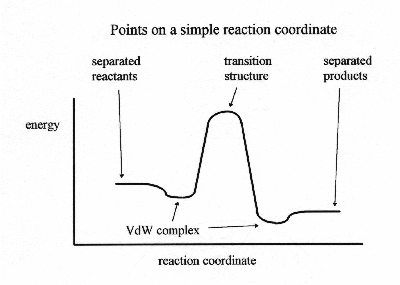
Different motions of a molecule will have different frequencies. As a general rule of thumb, bond stretches are the highest energy vibrations. Bond bends are somewhat lower energy and torsional motions are lower yet. The lowest frequencies are usually torsions between large pieces of large molecules and breathing modes in very large molecules. The simplest description of a vibration is a harmonic oscillator which describes springs exactly and pendulums with small amplitudes fairly well. A harmonic oscillator is defined by the potential energy being proportional to the square of the distance displaced from an equilibrium position. In a classical treatment of a vibrating object, the motion is fastest at the equilibrium position and comes to a complete stop for an instant at the turning points, where all of the energy is potential energy. The probability of finding the object is highest at the turning point and lowest at the equilibrium point. A quantum mechanical description of a harmonic oscillator uses the same potential energy function, but gives radically different results. In a quantum description, there are no turning points. There is some probability of finding the object at any displacement, but that probability becomes very small (decreasing exponentially) at large distances. The energy is quantized, with a quantum number describing each possible energy state and only certain energies possible. Very small objects, such as atomic particles behave according to the quantum description with low quantum numbers. Macroscopic objects under a quantum description will have very large quantum numbers with energy spacings that are too close together to measure and a probability distribution that becomes identical to the classical result in the limit of infinite quantum numbers. The fact that classical mechanics is a special case of quantum mechanics for large masses is called the "correspondence principle". The vibration of molecules is best described using a quantum mechanical approach. Molecules do not behave according to a harmonic oscillator description. Bond stretching is better described by a Morse potential and conformational changes have a sine wave type behavior. However, the harmonic oscillator description is used as an approximate treatment for low vibrational quantum numbers. The quantum mechanics equation (called the Schrodinger equation) has never been solved exactly for any chemical system containing more than one electron. However, many ways are known to approximate the solution. Approximation methods known as ab initio methods use mathematical approximations only. Frequencies computed with ab initio methods and a quantum harmonic oscillator approximation tend to be 10% too high (due to the difference between a harmonic potential and the true potential) except for the very low frequencies (below about 200 wave numbers) which are often quite far from the experimental values. Many studies are done using ab initio methods and multiplying the resulting frequencies by about 0.9 to get a good estimate of the experimental results. Semiempirical methods are another means of approximating the Schrodinger equation. In a semiempirical treatment, the computation is done much faster by neglecting part of the computation and using experimentally determined values to correct for the resulting errors. Vibrational frequencies from semiempirical calculations tend to be qualitative in that they reproduce the general trend mentioned in the second paragraph. However, the actual values are erratic. Some values will be close while others are too low or too high. The density-functional theory methods give frequencies with this same erratic behavior, but a somewhat smaller deviation from the experimental results. Some computer programs will output a set of frequencies containing six values near zero for the three degrees of translation and three degrees of rotation of the molecule. Other programs will use a more sophisticated technique to avoid computing these extra values, thus reducing the computation time. Before frequencies can be computed, the program must compute the geometry of the molecule since the normal vibrational modes are centered at the equilibrium geometry. When a negative frequency is computed, it indicates that the geometry of the molecule corresponds to a maximum of potential energy with respect to the positions of the nuclei. The transition state of a reaction is characterized by having one negative frequency. It is possible to compute vibrational frequencies using ab initio methods without using the harmonic oscillator approximation. For a diatomic molecule, the quantum harmonic oscillator energies can be obtained by knowing the second derivative of energy with respect to the bond length at the equilibrium geometry. For a non-harmonic oscillator energy, the entire bond dissociation curve must be computed, which requires far more computer time. Like wise, computing anharmonic frequencies for any molecule requires computing at least a sampling of all possible nuclear motions. Due to the enormous amount of time necessary to compute all of these energies, this sort of calculation is very seldom done. Another method for computationally describing molecules is called molecular mechanics. It is a non-quantum method in which the forces acting on the atoms are modeled as simple algebraic equations such as harmonic oscillators, Morse potentials, etc. All of the constants for these equations are usually obtained from experimental results. A set of equations and their constants is called a force field. A force field can be designed to describe the geometry of the molecule only or specifically created to describe the motions of the atoms. Calculation of the vibrational frequencies by determining the geometry then using a harmonic oscillator approximation can yield usable results if the force field was designed to reproduce the vibrational frequencies. NOTE: Many of the force fields in use today were not designed to reproduce vibrational frequencies in this manner. When using this method, there is not necessarily a 10% error between the results and the experiments, since the parameters may have been created by determining what harmonic parameters would reproduce the experimental results, thus building in the correction. As a general rule of thumb, mechanics methods do well if the compound being examined is similar to those used to create the parameters. Molecular mechanics does not do so well if the structure is significantly different from the compounds in the parameterization set. Another technique built around molecular mechanics is a dynamics simulation. In a dynamics simulation, the atoms move around for a period of time following Newton's equations of motion. This motion is a superposition of all of the normal modes of vibration so frequencies can not be determined directly from this simulation. However, the spectrum can be determined by doing a Fourier transform on these motions. The motion corresponding to a peak in this spectrum is determined by taking just that peak and doing the inverse Fourier transform to see the motion. This technique can be used to calculate anharmonic modes, very low frequencies and frequencies corresponding to conformational transitions. However, a fairly large amount of computer time may be necessary to get enough data from the dynamics simulation to get a good spectrum. Another related issue is the computation of the intensities of the peaks in the spectra. Peak intensities depend upon the probability that a particular wavelength photon will be absorbed or Raman scattered. These probabilities, can be computed from the wave function by first computing the transition dipole moment. Some types of transitions turn out to have a zero probability due to the molecules symmetry or the spin of the electrons. This is where spectroscopic selection rules come from. Ab initio methods are the preferred way of computing intensities. Although intensities can be computed using semiempirical methods, they tend to give rather poor accuracy results for many chemical systems. In conclusion it is possible to use computational techniques to gain insight into the vibrational motion of molecules. There are a number of computational methods available which are have varying degrees of accuracy and difficulty. These methods can be powerful tools if the user is aware of their strengths and weaknesses.
Further Information
The seminal text on molecular vibrations is
For an introductory level overview of computational chemistry see
An introduction to IR spectroscopy
Harmonic and anharmonic vibrations An expanded version of this article will be published in "Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems" by David Young, which will be available from John Wiley & Sons in the spring of 2001. |
[ CCL Home Page ]
[ topics ]
[ Raw Version of this page ]
| Modified: Thu Jan 4 02:29:24 2001 GMT |
| Page accessed 10875 times since Thu Jan 4 18:12:40 2001 GMT |
{kind=link}
{kind=link}
{kind=link}
{kind=link}