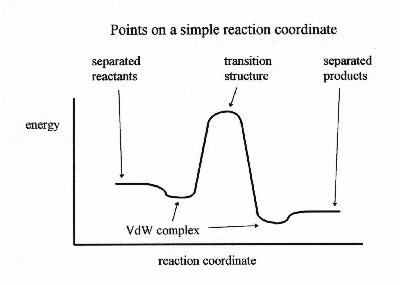
Short of determining an entire reaction coordinate, there are a number of structures and their energies that are important to defining a reaction mechanism. For the simplest single step reaction, there would be five of these structures.
A transition structure is mathematically defined as the geometry which has a zero derivative of energy with respect to moving every one of the nuclear coordinates and has a positive second derivative of energy for all but one geometric movement which has a negative curvature. Unfortunately, this description describes many structures other than a reaction transition such as an eclipsed conformation or the intermediate point in a ring flip or any structure with a higher symmetry than the ground state of the compound.
Predicting what a transition structure will look like (without the aid of a computer) is difficult for a number of reasons. Such a prediction might be made based on a proposed mechanism which is incorrect. The potential energy surface around the transition structure is often much more flat than the surface around a stable geometry ... thus there may be large differences in the transition structure geometry between two seemingly very similar reactions and with very small differences in energy.
Computationally it has been possible to determine transition structures for many years, although not always easy. Experimentally it has only recently become possible to examine reaction mechanisms directly using femtosecond pulsed laser spectroscopy. It will be some time before these techniques can be applied to all of the compounds that are accessible computationally. Further more, these techniques yield vibrational information rather than an actual geometry for the transition structure.
This technique has occasionally been applied to orbital based methods, where it goes under the name "seam searching". The rest of the techniques mentioned in this document are applicable to semiempirical, density functional theory (DFT) and ab initio techniques.
The best way to predict how well a given level of theory will describe a transition structure is to look up results for similar classes of reactions. Tables of such data are provided by Hehre in the reference listed below.
In this case the transition structure must have D3h symmetry with the two F atoms arranged axially and the H atoms equatorial. In fact, the transition structure is the lowest possible energy compound that satisfies this symmetry criteria.
In this case, the transition structure can be found by forcing the structure to have the correct symmetry then optimizing the geometry. This means geometry optimization rather than transition structure finding algorithms are used. This is a benefit because geometry optimization algorithms are generally more stable and reliable than transition structure algorithms.
For systems where the transition structure is not defined by symmetry it is often best to ensure that the starting geometry does not have any symmetry. This helps avoid converging to a solution which is an energy maximum of some other type such as an eclipsed conformation.
This is a quasi-Newton technique which implicitly assumes that the potential energy surface has a quadratic shape. Thus the optimization will only be able to find the correct geometry if the starting geometry is sufficiently close to the transition structure geometry to make this a valid assumption. Quasi-Newton techniques are generally more sensitive to the starting geometry than the synchronous transit methods discussed below. One good way to get a structure close to the correct transition structure is to use a transition structure from a very similar system (i.e. the same reaction with different functional groups).
Simplex optimizations have been tried in the past. These do not assume a quadratic surface, but require far more computer time and are seldom incorporated in commercial software. Due to the unavailability of this method to most researchers, it will not be discussed further here.
The optimization of a transition structure will be much faster using methods for which the Hessian can be analytically calculated. For methods which incrementally compute the Hessian (i.e. the Berny algorithm) it is advantageous to start with a Hessian from some simpler calculation, such as a semiempirical calculation.
When a transition structure is determined by starting from a single initial geometry, the calculation is very sensitive to the starting geometry. One excellent technique is to start with the optimized transition structure of another reaction which is expected to procede by the same mechanism then replace functional groups to give the desired reactants without changing the arrangement of the atoms where bonding is being changed. If no known transition structure is available, try setting the lengths of bonds being formed or broken intermediate to their bonding and van der Waals lengths. Often it is necessary for the starting geometry to have no symmetry (ignoring wave function symmetry is usually not sufficient).
The simplest way to guess the shape of a transition structure is to assume that each atom is directly between the position where it starts and the position where it ends. This linear motion approximation is called linear synchronous transit (LST). This is a good first approximation, but it has its failings. Consider the motion of an atom which is changing bond angle with respect to the rest of the molecule. The point half way between its starting and ending positions on the line connecting those positions will give a shorter than expected bond length and thus be (perhaps significantly) higher in energy.
The logical extension of this technique is the quadratic synchronous transit method (QST). These methods assume the coordinates of the atoms in the transition structure will lie along a parabola connecting the reactant and product geometries. QST generally gives some improvement over LST although it may be a very slight improvement.
Many programs allow the user to input a weighting factor (i.e. to give a structure that is 70% products and 30% reactants). This allows the application of the Hammond postulate that the transition structure will look more like the reactants for an exothermic reaction and more like the products for an endothermic reaction.
These techniques have been very useful for simple reactions, but have limitations. The down side is that each of these, even at their best is designed around the assumption that the reaction is a single step with a concerted motion of all atoms. For multi-step reactions, these techniques can be used individually for each step. For a reaction which has only one transition structure but the motion is not concerted (i.e. breaking one bond then forming another) it may be better to use starting geometries created by hand or eigenvalue-following.
There are distinct differences in the way these methods are implemented in specific software packages. Some software packages will require the user to choose a transit method to obtain a starting geometry then run a separate calculation with a quasi-Newton method. Other software packages will have an automated way of runing the transit method calculation followed by a quasi-Newton calculation. There have even been algorithms proposed for allowing the program to make decisions concerning which method to use at each step of the optimization.
Likewise a transition structure can be obtained by following the reaction path from the equilibrium geometry to the transition structure. This technique is known as eigenvalue-following because the user specifies which vibrational mode should lead to a reaction given sufficient kinetic energy. This is not the best way to obtain an IRC, nor is it the fastest or most reliable way to find a transition structure. However, it has the advantage of not making assumptions about concerted motions of atoms or what the transition structure will look like.
Another technique is to use a pseudo reaction coordinate. This can be quite a bit of work for the user and requires more computer time than most of the other techniques mentioned. However it has the advantage of being very reliable and thus will work when all other techniques have failed. A pseudo reaction coordinate is calculated by first choosing a geometric parameter intimately involved in the reaction (such as the bond length for a bond that is being formed or broken). A series of calculations is then run in which this parameter is held fixed at various values from those in the reactants to those in the products and all other geometric parameters are optimized. This does not give a true reaction coordinate but an approximation to it which matches the true reaction coordinate perfectly only at the equilibrium geometries and transition structure. Typically the highest energy calculation from this set is used as the starting geometry for a quasi-Newton optimization. In a few rare cases involving very flat potential surfaces the quasi-Newton optimization may still fail. In this case, the transition structure can be calculated to any desired accuracy (within the theoretical model) by finding the energy maximum by varying the chosen geometric parameter in successively smaller increments.
This type of calculation does reliably find a transition structure. However, it requires far more computer time than any of the other techniques. As such, this is really only done when the research requires obtaining a potential energy surface for reasons other than just finding the transition structure.
It is also always important to look at the transition structure geometry to make sure that it is the reaction transition and not the transition in the middle of a ring flip or some other unintended process. If it is not clear from the geometry, that the transition structure is correct, displaying an animation of the transition vibrational mode should make it very clear.
It is possible that a transition structure calculation will give two negative frequencies (a second order saddle point) or more. This gives a little bit of information about the potential energy surface but it is extremely unlikely that such a structure has any significant bearing on how the reaction occurs. This type of structure will often be found if the starting geometry was given a higher symmetry than the transition structure should have.
The simplest way to get a reaction rate is to use the activation energy in the Arrehenius equation. The preexponential factor can be obtained from experimental observations or some simple theoretical method such as the kinetic theory of gasses. To a first approximation the activation energy can be obtained by subtracting the energies of the reactants and transition structure. A readily obtained additional correction to these energies is obtained by the addition of the zero point vibrational energy.
Simply using the activation energy assumes that the only way a reaction occurs is along the intrinsic reaction coordinate. It would be more correct to consider that reactions may occur which go through a geometry very similar to the transition structure as well. Variational transition state calculations take this into account. These calculations may require using the vibrational frequencies for the transition structure, the entire reaction coordinate or the entire potential energy surface. These calculations can also take into account tunneling through the reaction barrier. These calculations can give good results, but are very sensitive to subtle details like using a mass weighted coordinate system to specify the geometry.
Dynamical studies can be done to examine how the path and orientation of approaching reactants affects the reaction rate. These studies often start with a potential energy surface which was obtained from ab initio calculations. The amount of work necessary to study a reaction with these techniques may be far more than the work done to get the potential energy surface, which was not a trivial task in itself.
A nice review with more detailed information and examples is
M. L. McKee, M. Page in "Reviews in Computational Chemistry, Volume IV"
K. B. Lipkowitz, D. B. Boyd, Eds. page 35, VCH Publishers (1993)
A nice discussion from the stand point of the potential energy surface
starts on page 240 of
A. R. Leach "Molecular Modelling Principles and Applications" Longman (1996)
For more information on synchronous transit methods see
C. Peng, H. B. Schlegel Israel Journal of Chemistry 33, 449 (1993)
Obtaining transition structures from molecular mechanics is discussed in
F. Jensen J. Comp. Chem. 15, 1199 (1994)
An expanded version of this article will be published in "Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems" by David Young, which will be available from John Wiley & Sons in the spring of 2001.