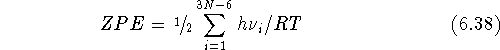By far, the most popular application of the empirical potential energy function
is to find the geometry
of a molecule (or an assemblage of molecules) which corresponds to a minimum
potential energy. The process of finding a minimum of an empirical
potential energy function is called frequently Molecular Mechanics (MM).
This process produces a motionless molecule of idealized geometry. In reality,
the molecules undergo thermal motions and constantly change their
geometry. Molecular Dynamics, (MD), and Monte Carlo, (MC) are
based on principles of statistical mechanics
and allow calculation of thermodynamic properties of molecular systems by
simulating these thermal fluctuations of geometry and corresponding energies.
These methods will be described briefly later in this chapter. For a review
of molecular mechanics consult Burkert & Allinger, (1982).
The potential energy function refers to a
ground electronic state of the molecule and does not explicitly incorporate
electronic structure. Hence, it cannot be used to study electronic spectra
or covalent bond formation/breaking. These phenomena are studied
with quantum methods. There are methods which combine a quantum description
for the small portion of the molecule (e.g., groups of substrate and enzyme
interacting in the active center) with a molecular
mechanics description for the rest of the molecule
(see e.g., Field et al., 1990).
The minimization of the potential energy function (i.e., geometry optimization)
involves a search for the minimum of a function, and, to be efficient,
requires calculations of derivatives of a function
(in this case, the potential
energy) versus independent variables (in our case, coordinates).
Most programs use cartesian coordinates as independent variables,
however, in some cases, internal coordinates may be used.
The derivatives of potential energy are denoted as:
where is the gradient (i.e., first derivative) of the potential energy
with respect to a cartesian coordinate
of an atom,
and is the second derivative of the energy with respect to
the cartesian coordinates. In most modern programs these derivatives are
calculated analytically, i.e., the appropriate mathematical formulae
for corresponding terms are incorporated into the program. Some older codes
compute derivatives numerically by approximating the slope
of an energy function (or its gradient in the case of second derivatives) from
finite differences.
The table of all possible second derivatives versus cartesian coordinates
of atoms has 3N rows and 3N columns
and is called a Hessian matrix. The derivatives are used not
only in function minimization but also yield forces acting on
atoms (from energy gradients) and normal modes of vibration (from the Hessian
matrix). There are three
major approaches to finding a minimum of a function of many variables:
Search Methods -- utilizes only values of the function itself.
They are usually slow and inefficient, but are very simple to
program, since deriving cumbersome formulas for derivatives is not
necessary. In spite of their inefficiency, the search algorithms are
infalliable and always find a minimum.
For this reason, they are often used as an initial step,
when the starting point in
optimization is far from the minimum. Another disadvantage of search
techniques is that they are very inefficient for a large number of
optimized variables and converge very slowly when the number of variables
is more then 10.
The most popular algorithm in this class is called SIMPLEX.
Gradient Methods -- utilizes values of a function and
its gradients.
These are currently the most popular methods in molecular mechanics.
They offer a much better convergence rate than search methods and do not
require a lot of computer memory (only 3N first derivatives are needed).
However, in some situations they fail to converge to a minimum.
The conjugated gradient algorithm is considered the most robust
in this class.
Newton Methods -- are the most rapidly converging algorithms
which require values of function, and its first and second
derivatives. The memory required for storing the Hessian matrix is proportional
to (i.e., prohibitive for large macromolecules). The
BFGS algorithm
is considered the most refined one.
In general, the minimization methods are iterative. They require on input
some initial estimate for the position of the minimum,
and provide a better estimate for the minimum as a result.
This corrected estimate is used as an input into the next
cycle (i.e., iteration) and the process is continued until there
is no significant improvement in the position of the minimum.
Figure 6.23: Local and global minima for a function of one
variable and
an example of a sequence of solutions: for a descent series minimization algorithm.
Most search methods and minimization methods using derivatives are the
descent series methods, i.e., each iteration results in a solution
which corresponds to a lower (or equal) value for the energy function:
As a consequence, these methods can only find the minimum closest
to the starting estimate and will never cross to a minimum (however deep)
if it is separated from the starting estimate by a maximum (however small).
This situation is schematically illustrated in Fig. 6.23 where arrows
indicate the direction of geometry optimization depending upon the
starting point. Fig. 6.23 is only a cartoon to illustrate the
behavior of descent series minimization methods. Geometry optimization of
real molecular systems (e.g. a protein with a drug molecule bound) involves
simultaneous optimization of 3N cartesian coordinates, i.e, sometimes
many thousands of variables. There is no
general way of finding a global minimum (i.e., the minimum corresponding to
the lowest possible value of the function). A different
initial geometry will usually lead to a different final minimum. Only on very
simple molecules will the single geometry optimization yield the global
minimum on the first trial. It cannot be overemphasized
that the result of a single minimization is usually a local
minimum, not a global one. To find a global minimum (or at least, to be
more confident about it) you have to perform many minimizations and use
different initial coordinates for each run.
There is another practical difficulty with contemporary molecular
mechanics programs. Most of them optimize the geometry in atomic cartesian
coordinates. To illustrate the problem let us join Alice on her journey
to the other side of the mirror. The appearance of the
potential energy function in cartesian space
is dramatically different from the one given by
internal coordinates. If we could
see a cartesian representation of a potential energy surface in, let us say,
fifty dimensions, the picture would look like the cortex of
a human brain --
lots of narrow tortuous valleys of similar depth. This is because low energy
paths for individual atoms are very narrow due to the presence of
hard bond stretching and angle bending terms. The low energy paths
correspond only to the rotation of groups or large portions of the molecule
as a result of varying torsional angles. In other words, the low energy paths
for atoms
are strongly correlated in cartesian space and atoms can move easily only
along narrow grooves. On the other hand, the
outlook of the potential energy surface is very different in the space of
internal coordinates. The surface looks like a valley surrounded by high
mountains. The high peaks correspond to stretching and bending terms,
and close van der Waals contacts, while the bottom of the valley represents
the torsional degrees of freedom. If you happen to start at the mountain tops
in the internal coordinate space, the
minimizer sees the bottom of the valley clearly from above.
If you are in the valley, you also see where the mountains are.
In cartesian space, the minimizer walks along the bottom of a narrow winding
channel which is frequently a dead-end.
In more scientific terms, all contemporary function minimization procedures
operate efficienty for functions close to quadratic (i.e., for two dimensions
a parabola, for three dimensions a boat like shape, for n dimensions:
)
and are inefficient for functions of other shapes. Assuming that bond
lengths and valence angles are already optimized and the goal of minimization
is to find the minimum corresponding to optimal values of torsional angles,
cartesian space is probably the worst possible. Look at equations
6.16, 6.27 and 6.9, which relate the torsional energy
to cartesian coordinates.
The dependance of the torsional energy on cartesian coordinates
is as far from a quadratic shape as it can be! Moreover, it has several
minima. There are also additional advantages to using minimization
in internal coordinate space. In this space there is a clear separation of
variables into the hard ones (i.e., those whose small changes produce
large changes in the function value) and soft ones (i.e., those whose
changes do not affect the function value substantially). During function
optimization in internal coordinates, the minimizer first optimizes
the hard variables and in the subsequent iterations ``cleans up'' the details
by optimizing the soft variables. In cartesian coordinate space all
variables are of the same type, i.e., hard or soft depending on
preference. Changing a cartesian coordinate of an atom results
in simultaneous change of bond lengths, valence angles and torsonal angles
associated with this atom, and distances to non-bonded atoms.
For this reason, the minimizer is only involved with details,
and therefore in the
last stages of the optimization all variables are as important as at the
beginning. The reason why most programs optimize geometry in
cartesian space is that it is not easy to derive and manage expressions for
derivatives of potential energy function in internal coordinates.
Note that the derivative of a van der Waals energy term between two atoms
versus the cartesian
coordinates of some third atom is zero. This derivative depends only
on the
cartesian coordinates of the two atoms involved. The situation is dramatically
different in internal coordinate space. Since the distance between
two atoms depends on all bond lengths, bond angles and torsional angles
on the path between two atoms, the derivative of the van der Waals term
for the two atoms will depend on all these variables. There are
matrix methods of calculating derivatives of distances and angles as
contributions from individual internal coordinates, and the final
derivative is calculated from the chain rule using these parameters. But as
you can see, the problem is far from trivial.
To avoid evaluation of derivatives, some programs use search techniques
like SIMPLEX when optimization
is requested in internal coordinate space. Since SIMPLEX is inefficient
for large numbers of variables, usually only torsional angles are
optimized by this technique. If the number of variables is too large,
then a block technique is used where only a portion of all independent
variables is optimized at a given stage. Another reason why
internal coordinates are not used routinely for geometry optimization
is that modern molecular mechanics programs are usually a portion of
a molecular mechanics/dynamics package. As will be shown later in this
chapter, molecular dynamics in coordinates
other than cartesian is a difficult problem, since the terms containing
generalized momenta and generalized coordinates in the Hamiltonian
cannot be separated. Molecular mechanics and dynamics
share large portions of the actual code, and hence there is a preference by
software authors to use the space which is common to both methods.
The potential energy of the molecule calculated from a well balanced
force field represents a strain in the molecule. Augmented
with bond/group equivalents and statistical mechanical corrections,
it can be used to estimate the heat of
formation of a compound (i.e., its molar enthalpy of formation,
). The quantity can be used
to compare the relative stability of
different compounds. In most cases however, you should assume that the
calculated potential energy incorporates some arbitrary component which
depends upon the types of atoms and covalent bonds in the molecule,
i.e., comparison of the energies calculated for
molecules with different numbers and/or types of atoms and bonds cannot be
rigorous. For this reason, potential energy will in most cases reliably
assess the difference in energy between conformers,
but will fail if you attempt to calculate the energetics of incorporating
a new functional group into the molecule. Molecular mechanics can also provide
the interaction energy, , of two molecules A and B as:
where , , and are
potential energies of the optimized complex, the optimized molecule A, and
the optimized molecule B;
respectively. Note that the type and number of atoms and covalent bonds
in the complex AB is equal to their sum in isolated molecules A and B,
and the arbitrary ``energy zero'' should cancel out in this case.
For this reason, the difference between interaction energies calculated for
different complexes, (i.e., ) is
the preferred method over direct comparison of the energies of different
complexes (i.e., ).
Potential energy functions can also be used to estimate contributions
from intramolecular vibrations to the vibrational free
energy and vibrational entropy. These quantities, and
contributions from translation and rotation of the molecule as a whole,
vary with temperature
and are main contributors to the thermodynamic functions such as enthalpy,
free energy, specific heat, etc., (see, e.g., Hill, 1960).
A well known approach is to use the frequencies, ,
corresponding to normal modes within harmonic approximation (i.e, to
derive them from a mass scaled Hessian matrix at energy minimum).
The expressions for relating classical vibrational contributions to Helmholtz
free energy, , internal
energy, , heat capacity at constant volume
,
and entropy of the nonlinear molecule, are derived
in many standard
textbooks for statistical mechanics (see e.g., McQuarrie, 1976):
where R, T, and h are the gas constant, the
absolute temperature, and Planck's
constant respectively; and N denotes the number of atoms in the molecule.
Frequently, these values are augmented with a
correction to account for vibrations at 0 K, which is of quantum origin,
by adding a zero-point energy (ZPE) to the
free energy and the internal energy :
The harmonic approximation is quite accurate
for isolated molecules. However, for complexes of two or more molecules
or systems containing the
solvent, the harmonic approximation breaks down, because the motion of
the individual
molecules with respect to each other is diffusional, i.e.,
not constrained by a skeleton of rigid covalent bonds. The potential of this
motion is different than the simple parabola of a harmonic oscillator and
contains multiple minima and maxima.
In this case, molecular dynamics or Monte Carlo approaches are more reliable
for estimation of thermodynamic functions.


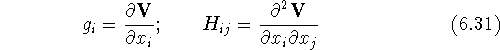
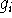 is the gradient (i.e., first derivative) of the potential energy
is the gradient (i.e., first derivative) of the potential energy
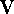 with respect to a cartesian coordinate
with respect to a cartesian coordinate 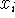 of an atom
of an atom
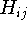 is the second derivative of the energy with respect to
the cartesian coordinates. In most modern programs these derivatives are
calculated analytically, i.e., the appropriate mathematical formulae
for corresponding terms are incorporated into the program. Some older codes
compute derivatives numerically by approximating the slope
of an energy function (or its gradient in the case of second derivatives) from
finite differences.
The table of all possible second derivatives versus cartesian coordinates
of atoms has 3N rows and 3N columns
and is called a Hessian matrix. The derivatives are used not
only in function minimization but also yield forces acting on
atoms (from energy gradients) and normal modes of vibration (from the Hessian
matrix). There are three
major approaches to finding a minimum of a function of many variables:
is the second derivative of the energy with respect to
the cartesian coordinates. In most modern programs these derivatives are
calculated analytically, i.e., the appropriate mathematical formulae
for corresponding terms are incorporated into the program. Some older codes
compute derivatives numerically by approximating the slope
of an energy function (or its gradient in the case of second derivatives) from
finite differences.
The table of all possible second derivatives versus cartesian coordinates
of atoms has 3N rows and 3N columns
and is called a Hessian matrix. The derivatives are used not
only in function minimization but also yield forces acting on
atoms (from energy gradients) and normal modes of vibration (from the Hessian
matrix). There are three
major approaches to finding a minimum of a function of many variables:
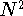 (i.e., prohibitive for large macromolecules). The
BFGS
(i.e., prohibitive for large macromolecules). The
BFGS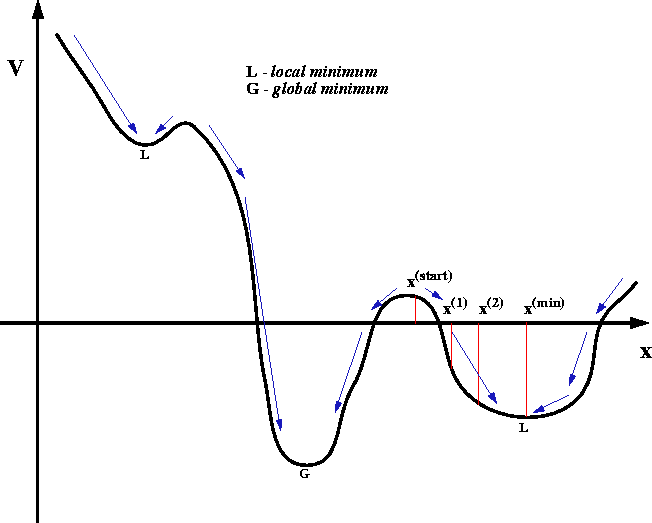
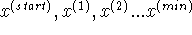 for a descent series minimization algorithm.
for a descent series minimization algorithm.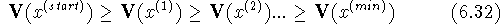
 )
and are inefficient for functions of other shapes. Assuming that bond
lengths and valence angles are already optimized and the goal of minimization
is to find the minimum corresponding to optimal values of torsional angles,
cartesian space is probably the worst possible. Look at equations
)
and are inefficient for functions of other shapes. Assuming that bond
lengths and valence angles are already optimized and the goal of minimization
is to find the minimum corresponding to optimal values of torsional angles,
cartesian space is probably the worst possible. Look at equations
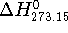 ). The quantity
). The quantity 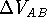 , of two molecules A and B as:
, of two molecules A and B as:
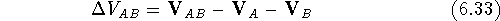
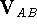 ,
, 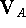 , and
, and 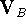 are
potential energies of the optimized complex, the optimized molecule A, and
the optimized molecule B;
respectively. Note that the type and number of atoms and covalent bonds
in the complex AB is equal to their sum in isolated molecules A and B,
and the arbitrary ``energy zero'' should cancel out in this case.
For this reason, the difference between interaction energies calculated for
different complexes, (i.e.,
are
potential energies of the optimized complex, the optimized molecule A, and
the optimized molecule B;
respectively. Note that the type and number of atoms and covalent bonds
in the complex AB is equal to their sum in isolated molecules A and B,
and the arbitrary ``energy zero'' should cancel out in this case.
For this reason, the difference between interaction energies calculated for
different complexes, (i.e., 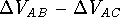 ) is
the preferred method over direct comparison of the energies of different
complexes (i.e.,
) is
the preferred method over direct comparison of the energies of different
complexes (i.e., 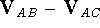 ).
).
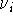 ,
corresponding to normal modes within harmonic approximation (i.e, to
derive them from a mass scaled Hessian matrix at energy minimum).
The expressions for relating classical vibrational contributions to Helmholtz
free energy,
,
corresponding to normal modes within harmonic approximation (i.e, to
derive them from a mass scaled Hessian matrix at energy minimum).
The expressions for relating classical vibrational contributions to Helmholtz
free energy, 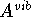 , internal
energy,
, internal
energy, 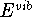 , heat capacity at constant volume
, heat capacity at constant volume
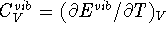 ,
and entropy
,
and entropy 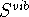 of the nonlinear molecule, are derived
in many standard
textbooks for statistical mechanics (see e.g., McQuarrie, 1976):
of the nonlinear molecule, are derived
in many standard
textbooks for statistical mechanics (see e.g., McQuarrie, 1976):
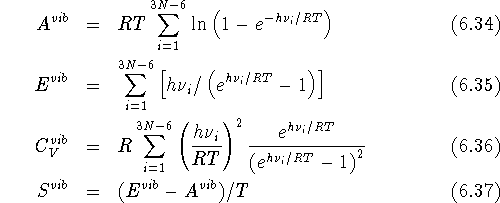
 K, which is of quantum origin,
by adding a zero-point energy (ZPE) to the
free energy
K, which is of quantum origin,
by adding a zero-point energy (ZPE) to the
free energy