Proton affinity 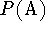 is defined as the negative of the molar enthalpy
change at 298.15 K (
is defined as the negative of the molar enthalpy
change at 298.15 K ( 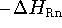 ) for the reaction
A + H
) for the reaction
A + H 
 AH , or in the case of anions,
A
AH , or in the case of anions,
A  + H AH.
To calculate these values from theory for gas phase reactions we may,
in most cases, obtain adequate results assuming ideal gas behaviour:
+ H AH.
To calculate these values from theory for gas phase reactions we may,
in most cases, obtain adequate results assuming ideal gas behaviour:
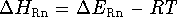 . Assuming that there is only
one conformer present, the energy E(T) of a mole
of gas consisting of nonlinear polyatomic molecules can be approximated as
. Assuming that there is only
one conformer present, the energy E(T) of a mole
of gas consisting of nonlinear polyatomic molecules can be approximated as 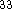 :
:
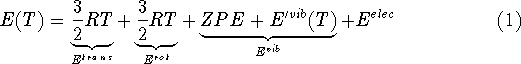
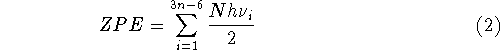
where n denotes the number of atoms in the molecule,
ZPE is the zero point energy, 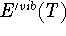 represents the
temperature dependent portion of vibrational energy,
represents the
temperature dependent portion of vibrational energy,
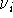 are the calculated vibrational frequencies and
are the calculated vibrational frequencies and
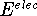 is the electronic energy
is the electronic energy  .
The change in the energy occuring during protonation
of 1 mole of gas at 298.15
.
The change in the energy occuring during protonation
of 1 mole of gas at 298.15  K,
K,
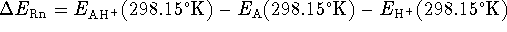 ,
will therefore consist
of the following components:
,
will therefore consist
of the following components:
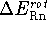 is nonlinear).
In our case, all parent and protonated molecules are nonlinear, hence,
is nonlinear).
In our case, all parent and protonated molecules are nonlinear, hence,
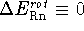 .
.
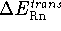
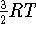 ,
the net
,
the net 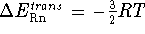 ; i.e.,
for 298.15 K
; i.e.,
for 298.15 K 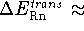 0.88873249 kcal/mol.
0.88873249 kcal/mol.
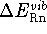
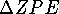 is significant.
The
is significant.
The 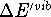 is usually much less then 1 kcal/mol
at room temperatures (i.e., much smaller than the experimental error)
and is included here only for completeness.
is usually much less then 1 kcal/mol
at room temperatures (i.e., much smaller than the experimental error)
and is included here only for completeness.
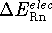
Combining the above, the following expression for proton affinity was used in actual computations:
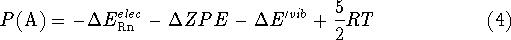
The values of and the vibrational frequences were obtained from
quantum calculations. Programs Gaussian90  and ACES2
and ACES2  were used for traditional ab initio calculations.
The original double-zeta basis set of Dunning/Huzinaga
were used for traditional ab initio calculations.
The original double-zeta basis set of Dunning/Huzinaga 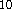 (9s,6p/4s)
(9s,6p/4s)  (6111,411/31) was augmented with
polarization functions:
d-type on C, N, O with exponents
equal to: 0.8, 0.8, and 0.9 respectively, and p-type with an
exponent equal to 1.0 for H. Two sets of ab initio calculations
were performed. DH6D set used cartesian gaussians (i.e., six d-type functions)
and the DH5D
(6111,411/31) was augmented with
polarization functions:
d-type on C, N, O with exponents
equal to: 0.8, 0.8, and 0.9 respectively, and p-type with an
exponent equal to 1.0 for H. Two sets of ab initio calculations
were performed. DH6D set used cartesian gaussians (i.e., six d-type functions)
and the DH5D  set used five d-type functions with the angular part
represented by spherical harmonics.
These DZP basis sets had similiar characteristics
to the basis sets we used
in our DFT calculations. The RHF and MP2 calculations were also performed
for formic acid and its anion with a DH6D basis set augmented
with diffuse s-type and p-type functions (with exponent 0.0845
set used five d-type functions with the angular part
represented by spherical harmonics.
These DZP basis sets had similiar characteristics
to the basis sets we used
in our DFT calculations. The RHF and MP2 calculations were also performed
for formic acid and its anion with a DH6D basis set augmented
with diffuse s-type and p-type functions (with exponent 0.0845 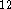 )
on oxygen atoms. It is well known that
adding diffuse functions on nonhydrogen atoms
dramatically improves results for proton affinities
)
on oxygen atoms. It is well known that
adding diffuse functions on nonhydrogen atoms
dramatically improves results for proton affinities  though
it is less important for molecular geometry.
This basis set is referred to later as DH6D
though
it is less important for molecular geometry.
This basis set is referred to later as DH6D  .
.
The ab initio results were compared with those obtained from DFT
calculations performed with the programs: DGauss  ,
DMol
,
DMol  and an academic version of deMon
and an academic version of deMon  .
The DGauss and deMon programs use the LSD potential developed by
Vosco et al.
.
The DGauss and deMon programs use the LSD potential developed by
Vosco et al.  while DMol incorporates the Barth and Hedin LSD
potential
while DMol incorporates the Barth and Hedin LSD
potential 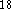 .
.
Basis sets of double-zeta quality with polarization functions were used in
all DFT programs. In DMol, the DNP (Double Numerical with Polarization)
basis set included with the program was selected. The basis sets in
this program are given numerically
as cubic spline functions. The 300 radial points spaning a range
from nucleus to an outer distance of 10 bohrs is a default.
The angular portion of each basis function
corresponds to the appropriate spherical harmonic.
The DGauss and deMon basis functions
are analogous to those widely used in traditional ab initio
calculations, i.e., they are represented analytically by a combination
of primitive gaussian functions; however, they have been reoptimized for the
DFT calculations 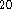 . For DGauss and deMon DZVP atomic basis set
and A2 auxiliary fitting set was used for 1st row atoms
and DZVPP/A1 set for hydrogens. The contraction pattern for the atomic
basis set was
(9s,5p,1d/5s,1p) (621,41,1/41,1). In other words, for 1st
row atoms there were three s-type basis functions combining six, two, and
one gaussian primitives, respectively; two sets of p-type basis functions
containing
four and one primitives, and a single primitive for each of the six d-type
polarization functions
. For DGauss and deMon DZVP atomic basis set
and A2 auxiliary fitting set was used for 1st row atoms
and DZVPP/A1 set for hydrogens. The contraction pattern for the atomic
basis set was
(9s,5p,1d/5s,1p) (621,41,1/41,1). In other words, for 1st
row atoms there were three s-type basis functions combining six, two, and
one gaussian primitives, respectively; two sets of p-type basis functions
containing
four and one primitives, and a single primitive for each of the six d-type
polarization functions 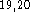 . For hydrogens two s-type
functions combining four and one gaussian primitives, and p-type polarization
functions consisting of one gaussian were used as a basis set.
. For hydrogens two s-type
functions combining four and one gaussian primitives, and p-type polarization
functions consisting of one gaussian were used as a basis set.
Beside atomic basis set, in DGauss and deMon, auxiliary sets of functions
are used to fit charge density and exchange-correlation potential .
For each atom there is one set for fitting density and another set for fitting
exchange-correlation potential. The sets consist of
uncontracted s-type gaussians and a few groups of s, p, and d-type cartesian
gaussians sharing the same exponent. The composition of these sets is denoted
as (A,B;C,D), where A,B pair specifies that A s-type gaussians, and B groups
of spd gaussians were used for fitting density, and C s-type gaussians and
D groups of spd gaussians were used to fit exchange-correlation potential.
The A2 auxiliary set was denoted (4,4;4,4), while the smaller A1 set used
for hydrogens was (3,1;3,1).
Starting geometries for molecules were obtained from model building with
Sybyl  using previous computational results
using previous computational results 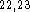 and experimental
data
and experimental
data 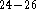 .
Geometries of protonated molecules were obtained by placing
a proton at position of the lone electron pair of the accepting atom.
The X-H bond lengths from the corresponding unprotonated molecules
were used for these new bonds as starting values.
These geometries were then fully optimized within given method.
It was absolutely necessary since these final geometries were
used for normal modes and vibrational frequency calculations.
Atom numbering used throughout this paper is shown in Figure 1 which
was produced with the MindTool software
.
Geometries of protonated molecules were obtained by placing
a proton at position of the lone electron pair of the accepting atom.
The X-H bond lengths from the corresponding unprotonated molecules
were used for these new bonds as starting values.
These geometries were then fully optimized within given method.
It was absolutely necessary since these final geometries were
used for normal modes and vibrational frequency calculations.
Atom numbering used throughout this paper is shown in Figure 1 which
was produced with the MindTool software 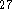 .
.
Ab initio geometry optimizations were
performed in DIRECT SCF mode with Gaussian90,
and the resulting geometries were used with ACES2 for
vibrational frequency and normal mode calculations.
ACES2 computes normal modes and frequencies from the analytical
Hessian for RHF and MP2 calculations.
We also performed geometry optimizations
at the MP2 level (full core) with Gaussian90 in an analogous manner,
followed by frequency
calculations with ACES2. In the MP2 case we used the RHF-optimized geometries
as starting data.
The DFT calculations with both DGauss and DMol were carried out with full geometry optimization at the Local Spin Density (LSD) level. Geometry optimizations with nonlocal gradient corrections were performed with deMon. Geometry optimization was followed by frequency calculations. For all DFT codes the Hessian needed for frequency and normal modes evaluation was calculated from analytical energy gradients by finite differences with a step of 0.01 bohr.
Integration grid in DGauss and deMon was of similar quality (MEDIUM and FINE respectively). In DMol the FINE integration grid density was used and the the maximum angular momentum number (LMAX) of the multipolar functions used to analitically fit electron density and exchange-correlation potential was 2 for hydrogens and 3 for other atoms.
Nonlocal density gradient corrections to the
LSD energies were not available in this version of DMol.
The version of DGauss used by us provided for single-point nonlocal
gradient corrections to LSD energies, while the deMon program
offered gradients of nonlocal gradient corrections,
i.e., provided for geometry optimization at this level.
Based on our previous experience 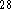 ,
we chose the Becke
,
we chose the Becke 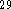 functional for the exchange
and Perdew
functional for the exchange
and Perdew 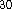 functional for the correlation potential,
which we label Becke-Perdew corrections.
The Becke-Perdew corrections were shown to improve results
compared to LSD approximation
functional for the correlation potential,
which we label Becke-Perdew corrections.
The Becke-Perdew corrections were shown to improve results
compared to LSD approximation 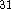 .
Since gradients of gradient-corrected energies could not be calculated
with this version of DGauss, single-point
corrected energies were calculated for the LSD-optimized geometries
and vibrational contributions to proton affinities were assumed to be the
same as for the LSD. For deMon, the fully-consistent LSD and NLSD results
are reported.
.
Since gradients of gradient-corrected energies could not be calculated
with this version of DGauss, single-point
corrected energies were calculated for the LSD-optimized geometries
and vibrational contributions to proton affinities were assumed to be the
same as for the LSD. For deMon, the fully-consistent LSD and NLSD results
are reported.