|
The rapid growth of available macromolecular structure data from X-ray
crystallography in the Protein Data Bank [Berstein 1977] has had
a large impact on computer-aided drug design and other computational
life science methodology. This structural data coupled with the
development of all-atom forcefields such as AMBER [Weiner 1986]
OPLS-AA [Jorgensen 1996] and MMFF94 [Halgren 1996] provides
means to conduct simulations of macromolecular structures such as
Energy Minimization, Molecular Dynamics and Ligand-Receptor Docking.
Explicit hydrogen atoms are required for all-atom molecular mechanics,
dynamics, docking and electrostatic calculations, and the initial state of
the hydrogen bond network and ionization state of titratable groups can
have a dramatic effect on simulations results. Unfortunately, most
macromolecular crystal structures contain little or no hydrogen coordinate
data due to limited resolution or disorder in the crystal.
Scientists that attempt to make use crystal structure data of macromolecules
for detailed simulations must spend a great deal of effort to prepare the
initial structure. Since the hydrogen data is missing in PDB files
most software programs assign default protonation states to the amino acid,
or nucleic acid residues as well as counter ions and other solvent. Such
assignments, at best, take only local structural details into account;
e.g., ensuring that a protonated nitrogen in the histidine sidechain
does not point at a ligated metal). Starting with such a default structure
(leaving aside the problem missing atoms and disorder) the protonation
uncertainties must be dealt with; commonly, one must determine
-
the rotamers of -SH -OH -CH3 and -NH3 groups in CYS, SER, TYR, THR, MET, LYS,
etc.;
-
the ionization states of acids and bases in ARG, ASP, GLU, LYS, HIS, etc.;
-
the tautomers of imidazoles (HIS) and carboxylic acids (ASP,GLU);
-
the protonation state of metal ligand atoms in CYS, HIS, ASP, GLU, etc.;
-
the ionization state of metals.
The addition of hydrogen atoms to a macromolecule is a non-trivial task. In
many cases, unambiguous assignments can be made (when backbone atoms are
involved) but the complexity of many hydrogen bond networks and ionic
interaction networks often makes protonation state determination very
difficult. Compounding the problem is the unfortunate fact that even the
heavy atoms of some chemical groups have uncertain identities; for example,
terminal amides in GLN and ASN and even the element identities of the
imidazole ring in HIS are often in question.
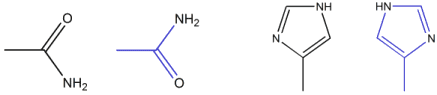
Consequently, one must add to the above list the determination of such
element identities. This is commonly referred to as flipping terminal
groups: the terminal amide groups in GLN, ASN and HIS can be left as read
from the PDB file (black) or "flipped" (blue). Possible
mis-assignments of element identities must also be considered in ligands
and additional groups must be examined (such as terminal sulfonamides
-SO2NH2).
 The protonation state problem exists not only with low resolution
X-ray structures, but also with high resolution structures. Hydrogen
coordinates are not always given for all atoms, and in some cases the
protonation assignment is questionable. For example, in the PDB
entry for a photoactive yellow protein (3PYP, 0.82 Å) one sees
that the deposited coordinates have a questionable proton assignment:
the phenol oxygen is deprotonated and the nearby carboxylate neutral and
has an unusual conformation (see figure to the right).
The protonation state problem exists not only with low resolution
X-ray structures, but also with high resolution structures. Hydrogen
coordinates are not always given for all atoms, and in some cases the
protonation assignment is questionable. For example, in the PDB
entry for a photoactive yellow protein (3PYP, 0.82 Å) one sees
that the deposited coordinates have a questionable proton assignment:
the phenol oxygen is deprotonated and the nearby carboxylate neutral and
has an unusual conformation (see figure to the right).
In other high resolution structures, one can see -CO2 groups
near ligands with no deposited hydrogen coordinates. In the
α-lytic protease structure (2H5C, 0.82 Å) such a
-CO2 group is adjacent to a glycerol molecule that has no
hydrogen atoms. In such a case, one cannot be certain that the
-CO2 is negatively charged, or neutral but with missing hydrogen
coordinates.
In general, the presence of a hydrogen atom in a PDB is clearly a statement
on the part of a crystallographer that a hydrogen was visible; however,
the absence of a hydrogen atom does not necessarily mean that a hydrogen
is definitely not there, merely that nothing was "visible".
Even if hydrogen coordinates are given for high resolution structures,
artifacts and phase errors of the electron density can lead to questionable
hydrogen coordinate assignments.
Computational attempts to deal with the macromolecular protonation state
assignment problem largely divide into two broad categories:
- Geometric Methods attempt to place protons often based upon local
hydrogen bonding environments or based upon optimization of hydrogen
bond networks. Crystallographic software often includes such procedures
(e.g., WHATIF).
- Electrostatic Methods attempt to place protons based upon
electrostatic field considerations and often attempt the calculation of
pKa shifts.
Geometric methods have the appealing feature that many proton assignments
can be made unambiguously from local hydrogen bonding environments -
sophisticated electrostatic treatment and dependence on partial charge
models and implicit solvent models is avoided.
The Reduce program [Word 1999] attempts to determine the
hydrogen coordinates using steric considerations and geometric hydrogen bond
network analysis; ionization and flipped states of HIS, GLU and ASP are
assigned on geometric grounds alone and rotamers are assigned from a fixed
discrete collection. In many cases, this program is effective; however,
since longer range electrostatic effects are taken into account,
Reduce can fail to make some clear assignments and simply cannot
properly assess complex ionic environments.
The electrostatic methods have appeal because of the more realistic
treatment of the underlying physics and thermodynamics. Computational
methods for calculating pKa values of residues often require sophisticated
electrostatic treatment using multiple Poisson-Boltzmann calculations
[Bashford 1992] followed by a search of the discrete ionization states
for the lowest free energy state [Gilson 1993]. Often, these programs
only assign the charge state and not the precise proton geometry. In
all-atom electrostatic methods, the protonation state of the non-titratable
groups is often taken to be some fixed default value, or assigned iteratively
taking into account the results of the titration calculation.
In all-atom methods, the protonation geometry of the non-titratable groups
depends on the ionization state of the titratable groups and vice versa.
Hence, the two problems must be solved simultaneously.
Were it not for the ionization state assignment (which changes the number
of particles in the system), forcefield methods, such as energy minimization
or molecular dynamics, could be used to determine flipped states and rotamers.
Unfortunately, the large number of local minima requires good initial
estimates of the hydrogen coordinates in order to properly search the
conformational space. Constant pH simulations (molecular dynamics or
Monte Carlo) are possible, but these methods require enormous computing time
and resources to properly converge.
The protonation assignment problem is inherently discrete; that is, it is
fundamentally a combinatorial search over a discrete set of states. Certainly,
the ionization state and tautomeric state can easily be thought of as
discrete. The rotamer problem can be made discrete by considering only
a fixed collection of rotamers (e.g., -OH dihedrals sampled every 30 degrees).
Thus, the rotamer, tautomer and ionization state calculation can be put into
a single context based upon discrete states. A particular protonation
state of a residue (or chemical group) consists of the 3D coordinates of all
of its hydrogens - tautomer and ionization states as well as a rotamer state.
For the amino acids, a reasonable collection of protonation states is
depicted below:
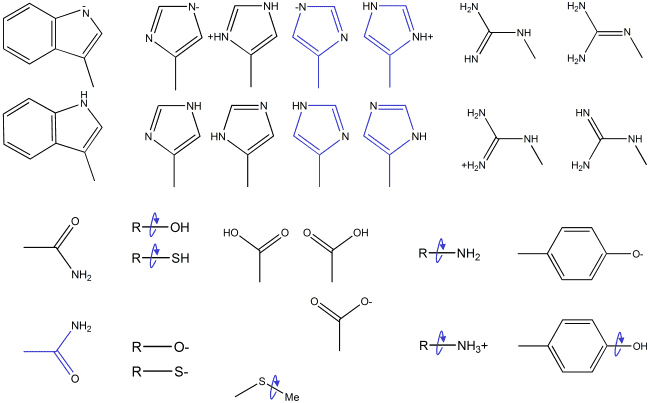
Here, all polar hydrogens are considered titratable, and terminal hydroxyls
and thiols are given discrete rotameric states (the number of which may
depend on hybridization or conjugation). Histidine has multiple
tautomers as well as "flipped" states, asparagine and glutamine
are given "flipped" states. The remaining amino acid sidechains
and the backbone can reasonably be assumed to exist in only one state
with reliably predictable hydrogen geometry (e.g., staggered for terminal
methyl in valine or isoleucine).
The foregoing discrete formulation of the protonation state assignment
problem is as follows: given the 3D coordinates of the heavy atoms of
a macromolecule, select a single protonation state (from the available
states) for each chemical group that is somehow "best". The
definition of "best" can be geometric or electrostatic depending
on the particular methodology and a complete method must specify the
definition of "best" as well as an algorithm for performing
the combinatorial search over the discrete state space.
In this document, we present a method - Protonate 3D - that solves the
discrete protonation state assignment problem. The method has been
implemented and is an application in the Molecular Operating Environment
in the 2007 release. In the following sections of this document, we present
the underlying theory and algorithms of the method as well as some results
when applied to PDB crystal structures. We summarize in the final section.
Protonate 3D solves the macromolecular protonation state assignment
problem by selecting a protonation state for each chemical group that
minimizes the total free energy of the system (taking titration into
account). The electrostatic interactions are approximated using the
Generalized Born / Volume Integral methodology [Labute 2007] with
a 15 Å cutoff. The discrete state search is performed using
an algorithm that solves the Unary Quadratic Optimization problem.
We now present the thermodynamic theory and algorithmic methodology that
lies at the heart of the Protonate 3D application.
The Protonate 3D methodology for protonation state assignment proceeds
as follows
- System Partition - The atoms of the system are partitioned into
separate chemical groups according to the patterns in a rule file. Each
chemical group will be given one or more topological states that include
tautomer states and ionization states. Each such group will have associated
with it a pKa and a strain energy used to penalize particular tautomeric
states. Each protonation state for each chemical group will subjected
to conformation generation to generate a collection of rotamers.
- Self Energy Calculation - The pKa values and strain energies along
with interactions to fixed (single state) chemical groups will be calculated
and stored in a vector, u.
- Interaction Matrix Calculation - The interaction energies between
each pair of 3D protonation states (corresponding to different chemical
groups) is calculated and stored in a matrix,
U.
- Dead End Elimination - A provably correct criterion is applied
in order to eliminate 3D protonation states that cannot appear in an optimal
solution. This reduces the complexity of the optimization procedure in
subsequent steps.
- System Optimization - A search for the particular configuration
of protonation states of the chemical groups that minimizes the quadratic
energy function
E(x) = xT U x / 2 +
uTx is performed. In this
function x is a binary vector that encodes the particular configuration
of selected protonation states (see below).
The discrete formulation of the protonation problem is very similar to the
sidechain placement and protein design problems with the protonation states
playing the role of the rotamer library popular in homology modeling
[Bower 1997]. Consequently, the methods used to solve these problems
can be applied to the protonation state problem [Looger 2001]
[Desmet 2002] [Canutescu 2003].
In the sidechain placement methods, each residue state (rotamer) is assigned
so that a pair-wise interaction function is optimized. Typically, a short
range steric function is used, such as the SCWRL function [Bower 1997].
Unfortunately, such functions are not accurate enough for titration
calculations since longer range electrostatic interactions, solvation
effects and titration free energies are not taken into account. Once
longer range effects are taken into account, the performance of algorithms
used in sidechain placement deteriorate - these algorithms rely on very
sparse pair-wise interactions. Nevertheless, the formalism is applicable
to the protonation state assignment problem provided that the interaction
function is suitable and that the search algorithm is capable of handling
the more numerous longer range interactions common to electrostatics
calculations.
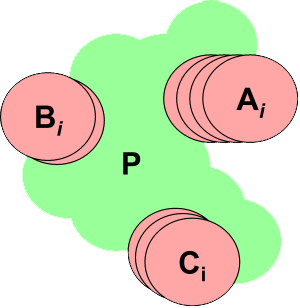 The partition of the system proceeds by (implicitly) cutting bonds connecting
sp3 carbons followed by the application of pattern matching
of patterns specified in a rule file. The results of the partition
are a collection of non-intersecting atom sets, each specifying a single
chemical group. Each such atom set will have an associated collection of
one or more ionization states, tautomeric strain energies, pKa values and
3D geometries (conformers).
The groups with exactly one state are assigned that single state and collected
into a single set of atoms P. Thus, the original system is divided
into a set of atoms P with a fixed protonation state, and one or
more chemical groups {A, B, C, ...} each
of which has two or more protonation states (including 3D geometry).
The partition of the system proceeds by (implicitly) cutting bonds connecting
sp3 carbons followed by the application of pattern matching
of patterns specified in a rule file. The results of the partition
are a collection of non-intersecting atom sets, each specifying a single
chemical group. Each such atom set will have an associated collection of
one or more ionization states, tautomeric strain energies, pKa values and
3D geometries (conformers).
The groups with exactly one state are assigned that single state and collected
into a single set of atoms P. Thus, the original system is divided
into a set of atoms P with a fixed protonation state, and one or
more chemical groups {A, B, C, ...} each
of which has two or more protonation states (including 3D geometry).
A binary vector x is used to encode a particular selection of
protonation states of {A, B, C, ...}
as follows. The bits of x are arranged in blocks, one for each
group with each block containing one bit for each particular configuration
of a particular chemical group. For example, if A has 4 protonation
states, B has 2 states and C has 3 states then the vector
x = [ 0 1 0 0 | 1 0 | 0 0 1 | ... ]
specifies the configuration of state 2 for A, state 1 for B
and state 3 for C. In this representation, a value of 1 in the
x vector selects a particular state and 0 de-selects a particular
state. Admissible values of the x are those in which there is
exactly one 1 bit in each block if bits that correspond to the states
of a particular chemical group. This constraint on the values of
x is called a unary constraint since the individual
states of the chemical groups are encoded in base 1 or unary notation.
Suppose that the interaction matrix U and self energy vector u
have been calculated as described above in the Self Energy and
Interaction Matrix steps of the procedure. (We will describe
the details of this matrix and vector below.) The optimization required
in the System Optimization step of the procedure is an instance
of Binary Quadratic Programming; the particular instance is called the
Unary Quadratic Optimization (UQO) problem, which is to optimize a
multi-dimensional quadratic function defined over binary vectors subject
to the unary constraint (that of selecting exactly 1 alternative in each block
of bits). More precisely, the UQO problem is
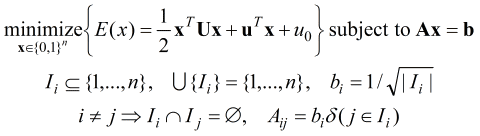
Here, the Ax = b condition enforces the unary constraint and the
x in {0,1}n enforces that x must be
an n-dimensional binary vector (n is the total number
of protonation states of A, B, C, ... Protonate 3D
uses the opt_UQO function in MOE to solve the UQO problem thereby
determining the minimum energy protonation state of the system. The
opt_UQO performs a very efficient recursive search through all of
the binary vectors x satisfying the unary constraint to locate the
vector x that minimizes the quadratic energy function E.
For protein-sized problems, the search can be typically conducted in under
one minute and often only a few seconds.
Prior to the recursive state search, an iterative procedure called
Dead End Elimination is applied in order to reduce the complexity
of the system. The Goldstein criterion [Goldstein 1994] is a condition
that can decide if a particular protonation state cannot possibly appear
in the optimal solution. Consider a particular chemical group
A with two particular protonation states r and s; if the
Goldstein criterion
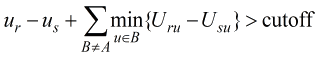
holds then protonation state r cannot possibly be within cutoff
energy units above the optimal solution. In the equation,
ur and us denote the self energies
of r and s respectively (elements of the u vector)
and Uij denotes the (i,j) entry of the
interaction matrix, U. The sum in the Goldstein criterion extends
over all other chemical groups, B, different from A.
The Protonate 3D application uses a cutoff of 0.
In the Dead End Elimination step of the procedure, the Goldstein
criterion is applied repeatedly to all pairs of protonation states for
all chemical groups until no eliminations are detected.
The Dead End Elimination step typically reduces the complexity of the
optimization problem dramatically and rapidly eliminates protonation states
that are obviously too high in energy (e.g., negatively charged tryptophan).
Configuration spaces with 10200 or so states are often reduced
to 1010 - hundreds of orders of magnitude.
It remains to describe the details of the interaction matrix U and self
energy vector u which define the energy function that is optimized
in the Protonate 3D calculation. Fundamentally, the energy function
is a free energy function - the number of particles in a titration
calculation changes and consequently, potential energy values of the
different configurations cannot be compared. The quadratic energy function
consists of the following terms:
- A van der Waals interaction energy;
- An electrostatic interaction energy;
- An implicit solvent interaction energy;
- A tautomeric strain energy;
- A rotamer strain energy;
- A titration free energy.
Each of these terms contributes (in some way) to the self energy vector,
u, or the interaction matrix, U. The tautomeric and rotamer
strain energies are inherent to each chemical group and contribute to the
vector, u. The tautomeric strain energy is taken from a rule file
which associates relative tautomer energies with the tautomers of each chemical
group. The rotamer strain energy is calculated when the conformations of
each chemical group are generated and is consistent with modern forcefields.
[Halgren 1996].
The van der Waals interactions play a relatively small role in the
Protonate 3D calculation; mostly they are used to distinguish
flipped states of terminal imidazoles and terminal amides, but sometimes
they may help distinguish correct protonation geometry [Word 1999].
The van der Waals interaction energies contribute both to the u vector
and U matrix. For each protonation state of a chemical group the van
der Waals interaction energy between the state and the fixed part of the
protein P is added to the u vector. For each pair for
protonation states of different chemical groups, the van der Waals interaction
energy is added to the appropriate element of the U matrix.
Protonate 3D uses an approximation to the Lennard-Jones 12-6 potential
energy function that mimics the repulsive potential and not the attractive
potential (which is more appropriate in implicit solvent model conditions).
The van der Waals
radii and well-depth parameters are those of the Engh-Huber forcefield
[Engh 1991] commonly used in crystallographic refinement.
The electrostatic energy terms are Coulombic in nature and make similar
contributions as the van der Waals interactions.
The electrostatic interaction energies contribute both to the u vector
and U matrix. For each protonation state of a chemical group the
electrostatic interaction energy between the state and the fixed part of the
protein P is added to the u vector. For each pair for
protonation states of different chemical groups, the electrostatic interaction
energy is added to the appropriate element of the U matrix.
The electrostatic functional form is configurable, and may be Coulomb's law,
a shifted-force Coulomb's law or a distance-dependent dielectric form of
Coulomb's law. For implicit solvent interaction energies, the electrostatic
energy used is always Coulomb's law. Partial charges are those of the
MMFF94 [Halgren 1996] forcefield (so that ligands can be handled).
The implicit solvent interaction energy and the titration free energy
form the basis of the titration calculation. Together with the pKa values
of the chemical groups and an input pH, free energies of titrations are
approximated with the following methodology. The fundamental implicit
solvent model that is used by Protonate 3D is the Generalized Born /
Volume Integral formalism [Labute 2007] with an additional salt
term:
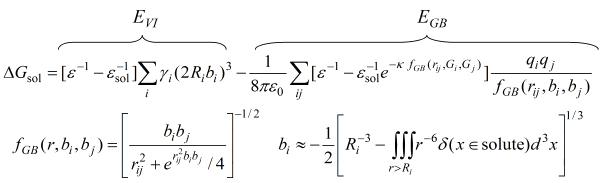
The GB/VI solvation free energy estimate consists of two parts, the
"self", or VI, part which calculates self energies of atoms
and the interaction, or GB, part, which estimates polarization interaction
energies between atoms. The inputs are fixed parameters (e.g., the
{Ri} and {γi}) and the
partial charges {qi} of the atoms and the salt concentration
(from which κ is derived).
Consider a titratable group AH in a protein environment P. In order
to estimate the free energy of the reaction PAH deprotonating to
PA- + H+ we introduce the thermodynamic cycle
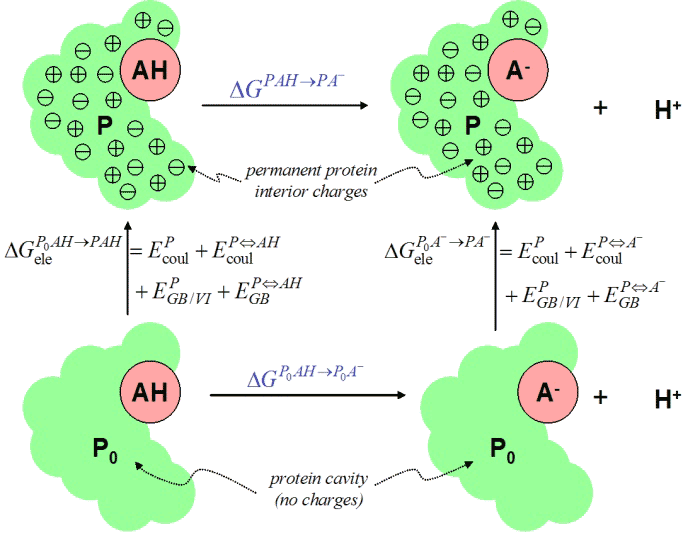
In this cycle, the charges within the fixed protein P are removed
(thermodynamically) and reintroduced along the vertical directions by
using specific interaction terms of the electrostatic and GB/VI energy
models. The symbol EXY denotes the interaction energy
between two sets of atoms X and Y.
The blue ΔG values on the horizontal paths denote unknown quantities.
The top value is the required free energy and the bottom horizontal value
arises from the pKa shift due to the protein cavity (without charges).
This bottom ΔG value can be estimated with another thermodynamic
cycle
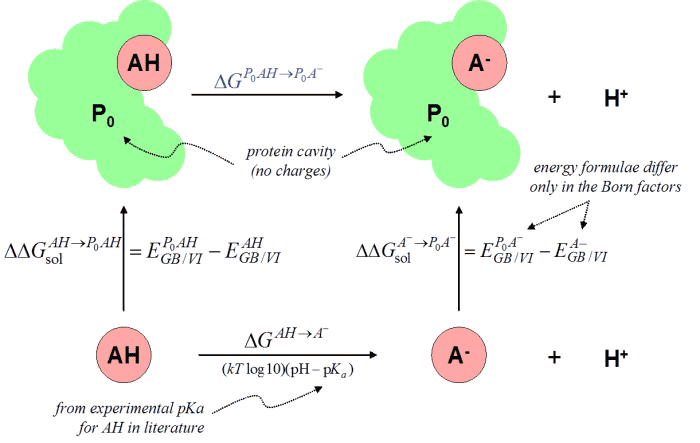
In this cycle, the protein cavity is removed and reintroduced along the
vertical directions. The vertical free energies are estimated, again,
from the GB/VI model and correspond to GB/VI calculations with different
Born factors (inversely proportional to the Born radii). Thus, the
previously unknown ΔG value (on top) is estimated with the vertical
free energy estimates and an experimental pKa value derived from the rule
file and the pH.
For polyprotic species (such as histidine) the above considerations generalize
to the following thermodynamic diagram.
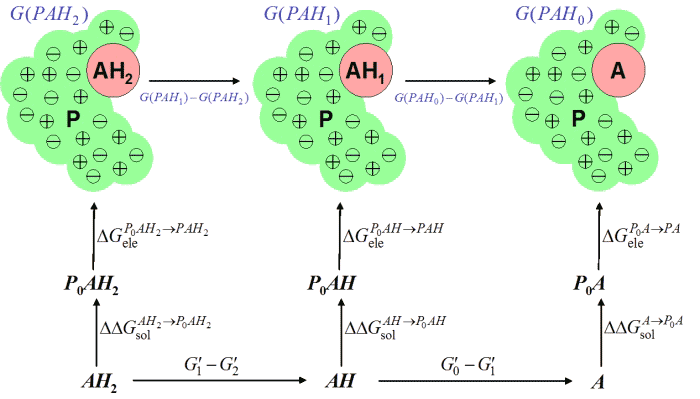
 in which the top horizontal ΔG values are the required titration
free energies of a titratable group A in situ in a protein
environment P. The bottom horizontal ΔG values are free
energy differences derived from the pKa values and the pH (as above).
The vertical ΔG values are those estimated from the GB/VI model
(as in the previous thermodynamic diagrams). The blue G values
are the free energies of the protonation states of the chemical group
A and have the property that their differences are equal to the
top horizontal ΔG values for the titration reactions; these values
will be added to the corresponding elements of the self energy vector
u required for the UQO formulation of the problem.
in which the top horizontal ΔG values are the required titration
free energies of a titratable group A in situ in a protein
environment P. The bottom horizontal ΔG values are free
energy differences derived from the pKa values and the pH (as above).
The vertical ΔG values are those estimated from the GB/VI model
(as in the previous thermodynamic diagrams). The blue G values
are the free energies of the protonation states of the chemical group
A and have the property that their differences are equal to the
top horizontal ΔG values for the titration reactions; these values
will be added to the corresponding elements of the self energy vector
u required for the UQO formulation of the problem.
The interaction matrix, U is populated with the GB interaction
energies between the various chemical group protonation states in a
straightforward way. Collecting all of the electrostatic, implicit
solvent and titration free energy values together, we arrive at the final
UQO energy function formulation
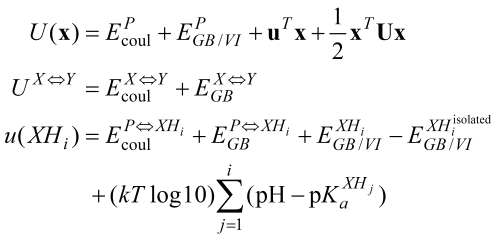

These values, together with the van der Waals contributions, the tautomeric
and rotamer strain energies form the complete free energy function E
required to compare different protonation state configurations of the entire
collection of chemical groups. This function E is optimized with the
opt_UQO function to produce optimal protonation state.
In this section we present some results of the Protonate 3D methodology
to certain PDB crystal structures.
 The PDB entry 2H5C (α-lytic protease) is a 0.82 Å crystal
structure with hydrogen atoms fit to the electron density.
The single protein chain has 198 residues and there are 2 glycerol molecules
and 11 SO4 ions.
The System Partition step produced 502 individual protonation states leading
to a U matrix of dimensions 502 by 502.
Prior to the Dead End Elimination step there were 10**61.8
system states and after the Dead End Elimination step there
were 10**6.1 states.
The Protonate 3D application required 17 seconds total run-time
on a 2 GHz Intel computer with 2 seconds spent in the UQO search.
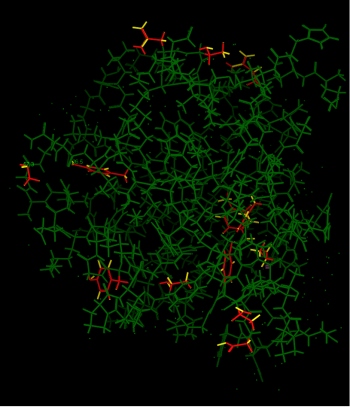
The output of Protonate 3D was compared to the crystal structure.
The PDB entry 2H5C (α-lytic protease) is a 0.82 Å crystal
structure with hydrogen atoms fit to the electron density.
The single protein chain has 198 residues and there are 2 glycerol molecules
and 11 SO4 ions.
The System Partition step produced 502 individual protonation states leading
to a U matrix of dimensions 502 by 502.
Prior to the Dead End Elimination step there were 10**61.8
system states and after the Dead End Elimination step there
were 10**6.1 states.
The Protonate 3D application required 17 seconds total run-time
on a 2 GHz Intel computer with 2 seconds spent in the UQO search.
The output of Protonate 3D was compared to the crystal structure.
The figure to the right shows the results of the comparison. Residues
without Protonate 3D protons that were in agreement with the
crystal structure are shown in green; disagreements are in red.
Agreement was designated if the ionization states were equal, the
tautomer and/or flip state was in agreement and the hydrogen rotamer
was within 60 degrees.
85% of all residues had their hydrogen rotamers within 15 degrees of the
crystal structure and 90% were within 60 degrees. All of the disagreements
were on the surface. Of the residues showing disagreement, 10 were
deposited without hydrogens - 7 serines, 2 threonines and 1 leucine.
There were two terminal amide disagreements, ASN60 and GLN223, both of which
were on the surface and had very little contact with the rest of the protein.
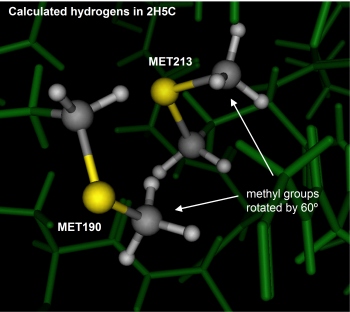
Two interacting methionine residues showed more than 60 degrees difference
between the prediction and the crystal structure (depicted below). The
terminal methyls were allow free rotation (no rotamer strain energy was
used and as a result they adopted eclipsed-like conformations. Perhaps
the addition of a rotation penalty (to model the rotation barrier) would
have resulted in staggered conformations. In any event, this disagreement
is quite minor; indeed, almost insignificant.
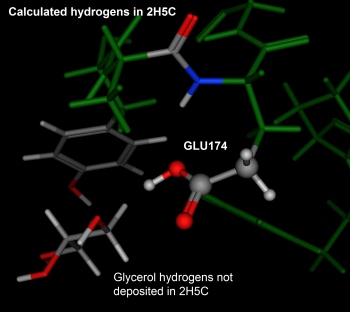
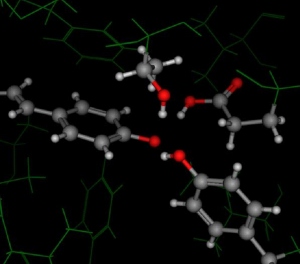
The figure to the right of the interacting methionine residues is a close-up
of GLU174 (mentioned in the introduction of this document). The terminal
carboxylic acid is adjacent to a glycerol molecule and neither the
carboxylic acid nor the glycerol showed hydrogen coordinates in the PDB
entry. Protonate 3D assigned a neutral state to GLU174. While this
is a formal disagreement we do not deem it serious.
 The PDB entry 3PYP (photoactive yellow protein) is a 0.82 Å
crystal structure with hydrogen atoms fit to the electron density.
The single protein chain has 125 residues with a covalent ligand.
The System Partition step produced 415 individual protonation states leading
to a U matrix of dimensions 415 by 415.
Prior to the Dead End Elimination step there were 10**46.1
system states and after the Dead End Elimination step there
were 10**4.1 states.
The Protonate 3D application required 5.2 seconds total run-time
on a 2 GHz Intel computer with 1.6 seconds spent in the UQO search.
The output of Protonate 3D was compared to the crystal structure.
The PDB entry 3PYP (photoactive yellow protein) is a 0.82 Å
crystal structure with hydrogen atoms fit to the electron density.
The single protein chain has 125 residues with a covalent ligand.
The System Partition step produced 415 individual protonation states leading
to a U matrix of dimensions 415 by 415.
Prior to the Dead End Elimination step there were 10**46.1
system states and after the Dead End Elimination step there
were 10**4.1 states.
The Protonate 3D application required 5.2 seconds total run-time
on a 2 GHz Intel computer with 1.6 seconds spent in the UQO search.
The output of Protonate 3D was compared to the crystal structure.
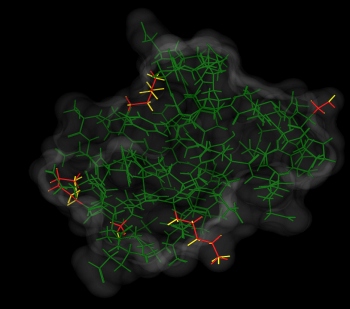
The figure to the right shows the results of the comparison. Residues
without Protonate 3D protons that were in agreement with the
crystal structure are shown in green; disagreements are in red.
Agreement was designated if the ionization states were equal, the
tautomer and/or flip state was in agreement and the hydrogen rotamer
was within 60 degrees.
The four active site residues showing disagreement were referred to in
the introduction of this document. The deposited coordinates showed
a deprotonated phenol in close proximity to a protonated GLU46 (see
the leftmost figure below). Moreover, the conformation of the GLU46
is unusual with the hydrogen in a kind of
trans conformation to the carbonyl oxygen. The nearby THR50
and TYR42 are donating hydrogen bonds to the (anionic?) phenol oxygen.
The deposited protonation state is largely questionable because of the
relative pKa difference between phenol and carboxylic acid; one would
expect that given the close proximity, the carboxylic acid of GLU46
would be deprotonated and the phenol of the covalent ligand be protonated
yet this is not the case in the crystal structure.
The rightmost figure below depicts the same residues with the protonation
state assigned by the Protonate 3D procedure.
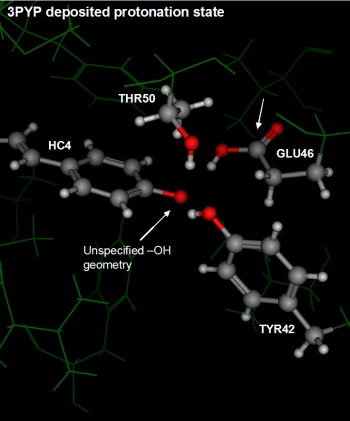 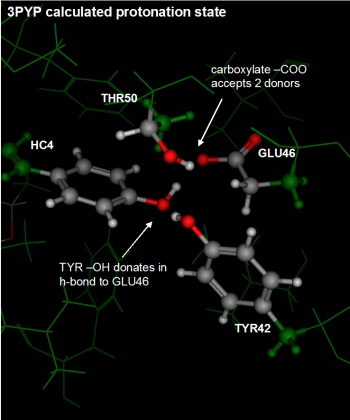
Protonate 3D protonated the phenol of the covalent ligand and
deprotonated the carboxylic acid of GLU46. The nearby THR50 and TYR42
are able to form favorable polar interactions with the THR50 now donating
is hydrogen to the GLU46.
 The PDB entry 1YK4 (electron transport rubredoxin 24L/R5S) is a
0.69 Å crystal structure with hydrogen atoms stated to
by "visible" in the PDB file header.
The single protein chain has 53 residues and one iron atom.
The System Partition step produced 189 individual protonation states leading
to a U matrix of dimensions 189 by 189.
Prior to the Dead End Elimination step there were 10**22
system states and after the Dead End Elimination step there
were 10**0.1 states.
The Protonate 3D application required 1.8 seconds total run-time
on a 2 GHz Intel computer with 0.8 seconds spent in the UQO search.
The output of Protonate 3D was compared to the crystal structure.
The PDB entry 1YK4 (electron transport rubredoxin 24L/R5S) is a
0.69 Å crystal structure with hydrogen atoms stated to
by "visible" in the PDB file header.
The single protein chain has 53 residues and one iron atom.
The System Partition step produced 189 individual protonation states leading
to a U matrix of dimensions 189 by 189.
Prior to the Dead End Elimination step there were 10**22
system states and after the Dead End Elimination step there
were 10**0.1 states.
The Protonate 3D application required 1.8 seconds total run-time
on a 2 GHz Intel computer with 0.8 seconds spent in the UQO search.
The output of Protonate 3D was compared to the crystal structure.
The figure to the right shows the results of the comparison. Residues
without Protonate 3D protons that were in agreement with the
crystal structure are shown in green; disagreements are in red.
Agreement was designated if the ionization states were equal, the
tautomer and/or flip state was in agreement and the hydrogen rotamer
was within 60 degrees.
85% of the residues had hydrogen rotamers within 10 degrees of the crystal
structure and 91% had hydrogen rotamers with 20 degrees of the crystal
structure. Three residues with 60 degree differences were protonated
lysines. All disagreements were on the surface. Only one residue showed
more than a 60 degree rotamer disagreement, a serine on the surface.
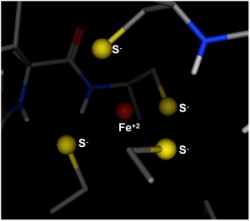 The 1YK4 structure has an iron surrounding by four CYS sulfurs. In the,
Protonate 3D procedure, the iron was allowed to adopt one of three
ionization states: +1, +2 or +3 without any redox penalty; in other words,
the remainder of the system determined the ionization state of the iron.
The figure on the right shows the output of the Protonate 3D procedure
at the iron center. The four CYS sulfurs are assigned anionic states
and the iron is assigned a +2 state, giving an overall -2 charge for the
environment. This assignment is quite reasonable, although one must
keep in mind that only a simplistic treatment of metal ligation was
conducted (entirely consisting of Coulombic and implicit solvent effects).
The 1YK4 structure has an iron surrounding by four CYS sulfurs. In the,
Protonate 3D procedure, the iron was allowed to adopt one of three
ionization states: +1, +2 or +3 without any redox penalty; in other words,
the remainder of the system determined the ionization state of the iron.
The figure on the right shows the output of the Protonate 3D procedure
at the iron center. The four CYS sulfurs are assigned anionic states
and the iron is assigned a +2 state, giving an overall -2 charge for the
environment. This assignment is quite reasonable, although one must
keep in mind that only a simplistic treatment of metal ligation was
conducted (entirely consisting of Coulombic and implicit solvent effects).
Generally, transition metals are treated in Protonate 3D in a disconnected
ionic manner. For example, zinc atoms are disconnected from their (possibly)
ligated atoms and their ionization states are selected from a fixed collection.
The possible ligated atoms are titrated in the usual manner. For this
reason, we allow histidine to adopt a negative charge state; this allows
for possible metal ligation of the otherwise neutral histidine.
For the most part, this (simplistic) metal treatment produces reasonable
results which are good starting points for further analysis, possibly with
a higher level of theory.
We have presented a method - Protonate 3D - for adding protons to
macromolecules. Tautomerism, titration, metal ligation, steric and
(implicit solvent) electrostatic interactions are addressed in a
thermodynamically sound manner. The combinatorial optimization is conducted
with an algorithm that solves the Unary Quadratic Optimization problem.
The method shows good agreement with (very) high resolution crystal structures
subject to some key limitations. Metal centers are handled by overly-simple
method (Coulombic). The method is sensitive (at times) to crystallographic
coordinates. Borderline titration cases may be unreliable due to the static
Generalized Born formalism in which calculation is conducted.
Protonate 3D has been implemented in the SVL programming language and
is available as part of the Molecular Operating Environment (2007 version).
| [Bashford 1992]
|
Bashford, D., Gerwert, K.; "Electrostatic Calculations of pKa Values of Ionizable Groups in
Bacteriorhodopsin".
J. Mol. Biol. 224 (1992) 473-486.
|
| [Berstein 1977]
|
Berstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer Jr., E. F.,
Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T., Tasumi, M.; "The Protein Data Bank: A Computer-Based Archival File for
Macromolecular Structures", J. Mol. Biol. 112 (1977) 535-542.
|
| [Bower 1997]
|
Bower, M. M., Cohen, F. E., Dunbrack Jr., R. L.; "Prediction of Protein Side-chain Rotamers from a Backbone Dependent Rotamer
Library: A New Homology Modeling Tool".J. Mol. Biol. 267 (1997) 1268-1282.
|
| [Canutescu 2003]
|
Canutescu, A. A., Shelenkov, A. A., Dunbrack Jr., R. L.;
A Graph-Theory Algorithm for Rapid Protein Side-chain Prediction
Protein Science 12 (2003) 2001-20014.
|
| [Desmet 2003]
|
Desmet, J., Spriet, J., Lasters, I.; "Fast and Accurate Side-Chain Topology and Energy Refinement (FASTER)
as a New Method for Protein Structure Optimization".PROTEINS: Structure, Function and Genetics 48 (2002) 31-43.
|
| [Engh 1991]
|
Engh, R. A., Huber, R.; "Accurate Bond and Angle Parameters for X-ray Protein Structure Refinement".
Acta Cryst. A47 (1991) 392-400.
|
| [Gilson 1993]
|
Gilson, M. K.; "Multiple-Site Titration and Molecular Modeling: Two Rapid Methods for
Computing Energies and Forces for Ionizable Groups in Proteins". PROTEINS: Structure, Function and Genetics 15 (1993) 266-282.
|
| [Goldstein 1994]
|
Goldstein, R. F.; "Efficient Rotamer Elimination Applied to Protein Side Chains
and Related Spin Glasses". Biophys. J. 66 (1994) 1335-1340.
|
| [Halgren 1996]
|
Halgren, T. A.; "The Merck Force Field".J. Comp. Chem. 17 (1996) 490-641.
|
| [Jorgensen 1996]
|
Jorgensen W. L., Maxwell, D. S., Tirado-Rives, J.; "Development and Testing of the OPLS All-Atom Force Field on Conformational
Energetics and Properties of Organic Liquids". J. Am. Chem. Soc. 117 (1996) 11225-11236.
|
| [Labute 2007]
|
Labute, P.; "The Generalized Born / Volume Integral (GB/VI) Implicit Solvent Model:
Estimation of the Free Energy of Hydration Using London Dispersion Instead
of Atomic Surface Area". J. Comp. Chem. (2007) - in press.
|
| [Looger 2001]
|
Looger, L. L., Hellinga, H. W.; "Generalized Dead-end Elimination Algorithms Make Large-scale Protein
Side-chain Structure Prediction Tractable: Implications for Protein
Design and Structural Genomics."
|
| [Word 1999]
|
Word, M. M., Lovell, S. C., Richardson, J. S., Richardson, D. C.; "Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of
Side-chain Amide Orientation". J. Mol. Biol. 285 (1999) 1735-1747.
|
| [Weiner 1987]
|
Weiner, S. J., Kollman, P. A., Nguyen, D. T., Case, D. A; "An All Atom Force Field for Simulations of Proteins and Nucleic Acids".
J. Comput. Chem. 7 (1986) 230.
|
|