|
pass
|
|
Delta.gif,
Image1.gif,
Image10.gif,
Image11.gif,
Image12.gif,
Image14.gif,
Image15.jpg,
Image2.gif,
Image3.gif,
Image4.gif,
Image5.gif,
Image6.gif,
Image7.gif,
Image8.gif,
Image9.gif,
README,
fig3.jpg,
fig4.jpg,
fig5.jpg,
fig6.jpg,
mailbox.gif,
overview.html,
pass.pdf,
pass10_2.0.36linux.tar.Z,
pass10_2.0.36linux.tar.gz,
pass10_sgi.tar.Z,
pass10_sgi.tar.gz,
pass10_sgi_n32mips3.tar.Z,
pass10_sgi_n32mips3.tar.gz,
pass10_sun.tar.Z,
pass10_sun.tar.gz,
pass_jcamd.html,
pass_usage.html,
passlogo.gif,
redline.gif,
redwhite.gif,
sigma.gif
|
|
|
I wish to submit the program "PASS" to the CCL as
SunOS,SGI, and Linux executables. I have uploaded tar files
containing both the executable and a "readme" file, which
explains PASS (shown below). I hope people find it useful.
Thanks.
Pat Brady
****************************************************************
readme - Release 1.0 of PASS ("Putative Active Sites with Spheres")
G. Patrick Brady, Jr., Ph.D.
DuPont Pharmaceuticals Co.
G.Patrick.Brady@dupontpharma.com
PASS web page can be reached via the Protein Data Bank (i.e. the
RCSB) at http://rcsb.rutgers.edu/pdb/software-list.html
This is freeware, and no guarantees are made or implied
regarding the performance of PASS.
--------------------------------------------------------------------
INSTALLATION
Only executables are provided, so installation amounts to simply
decompressing and unarchiving the appropriate file:
"gunzip filename.gz" then "tar -xvf filename.tar"
or "uncompress filename.Z" then "tar -xvf filename.tar"
pass10_sgi : SGI Irix 6.5
pass10_sgi_n32mips3 : SGI Irix (-n32 -mips3 compiler options)
pass10_sun : SunOS 5.6
pass10_linux : Linux
--------------------------------------------------------------------
SUMMARY
PASS (Putative Active Sites with Spheres) is a simple computational tool
that uses geometry to characterize regions of buried volume in proteins
and to identify positions likely to represent binding sites based upon
the size, shape, and burial extent of these volumes. PASS'S utility as
a predictive tool for binding site identification is tested by predicting
known binding sites of proteins in the PDB using both complexed macromolecules
and their corresponding apo-protein structures. The results indicate that
PASS can serve as a front-end to fast docking. The main utility of PASS
lies in the fact that it can analyze a moderate-size protein (~ 30 kD)
in under twenty seconds, which makes it suitable for interactive molecular
modeling, protein database analysis, and aggressive virtual screening efforts.
As a modeling tool, PASS (i) rapidly identifies favorable regions of the
protein surface, (ii) simplifies visualization of residues modulating binding
in these regions, and (iii) provides a means of directly visualizing buried
volume, which is often inferred indirectly from curvature in a surface
representation. PASS produces output in the form of standard PDB files,
which are suitable for any modeling package, and provides script files
to simplify visualization in Cerius2, InsightII, MOE, Quanta, RasMol,
and Sybyl. PASS is freely available to all.
--------------------------------------------------------------------
RUNNING PASS
PASS is run from the unix command-line; executing "pass" with no
arguments displays the syntax and a list of options (shown at the
bottom of this file).
--------------------------------------------------------------------
VISUALIZATION USING PASS-GENERATED SCRIPT FILES
Cerius2 (tested on v.3.8):
"Utilities -> Playback Script" then select the script, "filename.log"
InsightII (tested on v.98.0):
"File -> Source_File" then select the script file, "filename.bcl"
MOE (tested on v.1999.05):
In the MOE command window, type "'load filename.svl'" (with the
apostrophies) then "pass[]"
Quanta (tested on v.97.1003):
From the unix command-line, type "quanta -i filename.rec"
Or, while in Quanta, "Restart Quanta", then
"Replay Session -> Start" and select the script, "filename.rec"
RasMol (tested on v.2.6):
At the command window prompt, type "script filename.ras"
Sybyl (tested on v.6.5):
"File -> Take Commands" then select the script, "filename.col"
--------------------------------------------------------------------
TROUBLESHOOTING
Failure of PASS to run usually means either that (1) the executable
is incompatible with the machine on which it is being run, or (2)
there is something nonstandard in the input PDB file (e.g. multiple
occurrences of poorly resolved residue fragments- they are preceded
with letters, which must be removed and only one copy of the
residue retained).
Please report bugs, comments, and requests to g.patrick.brady@dupontpharma.com.
======================================================================
======================================================================
==== PASS - Putative Active Sites with Spheres ======================
============================================= G. Patrick Brady =======
======================================================================
= USAGE: pass ProteinPDBfile =
= =
= where include: =
= =
= <-outdir directory_path> ... Specifies that output (i.e. =
= visualization scripts, PDB files for probes, =
= ASPs, ligands, etc) is to be sent to the =
= specified directory path (defaults to the =
= current directory, ./ ) =
= <-more> ... Identify an enhanced set of probe spheres and =
= active site points (ASPs) =
= <-allprobes> ... Produce a display of all probe spheres =
= (i.e. suppress smoothing which, by default, =
= eliminates any probes that do not have at =
= least smoothcount neighbors lying within =
= smoothradius Angstroms). =
= <-volumes> ... Smooth the final probe spheres, group them =
= via clustering, and compute the volumes of the =
= resulting regions. Outputs several measures =
= of nearness to ligands, in the event that some =
= are present. =
= <-noprobes> ... Do NOT produce a PDB file of all final =
= probe spheres (default is to make this file) =
= <-layers> ... Produce a PDB file of each layer of probe =
= spheres (by default these files are NOT made) =
= <-waters> ... Treat waters as part of the protein (by =
= default, waters are removed and ignored) =
= <-heavyonly> ... Exclude hydrogen atoms from calculation =
= of probes. Default is to sense from input PDB =
= <-hydrogen> ... Include hydrogen atoms in probe calculation =
= <-ligand PDBfilename> ... Read-in a ligand from a =
= separate PDB file (supercedes any HETATM =
= ligand(s) in ProteinPDBfile) =
= Visualization Script Files: load, color, and render the protein, =
= probe spheres, ASPs, and ligand(s). In some cases =
= radial subsets centered on the ASPs are automatically =
= defined. =
= <-cerius2> ... Produce a Cerius2 script (filename.log) =
= <-insightII> ... Produce an InsightII script (filename.bcl) =
= <-moe> ... Produce a MOE script (filename.svl) =
= <-quanta> ... Produce a Quanta script (filename.rec) =
= <-rasmol> ... Produce a RasMol script (filename.ras) =
= <-sybyl> ... Produce a Sybyl script (filename.col) =
======================================================================
|


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
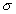{kind=link}