|
pass
|
|
Delta.gif,
Image1.gif,
Image10.gif,
Image11.gif,
Image12.gif,
Image14.gif,
Image15.jpg,
Image2.gif,
Image3.gif,
Image4.gif,
Image5.gif,
Image6.gif,
Image7.gif,
Image8.gif,
Image9.gif,
README,
fig3.jpg,
fig4.jpg,
fig5.jpg,
fig6.jpg,
mailbox.gif,
overview.html,
pass.pdf,
pass10_2.0.36linux.tar.Z,
pass10_2.0.36linux.tar.gz,
pass10_sgi.tar.Z,
pass10_sgi.tar.gz,
pass10_sgi_n32mips3.tar.Z,
pass10_sgi_n32mips3.tar.gz,
pass10_sun.tar.Z,
pass10_sun.tar.gz,
pass_jcamd.html,
pass_usage.html,
passlogo.gif,
redline.gif,
redwhite.gif,
sigma.gif
|
|
|
PASS Homepage

Putative Active Sites with Spheres
Keywords: protein active site, binding site, cavity detection, buried volume,
molecular modeling, computer-aided drug design
Summary
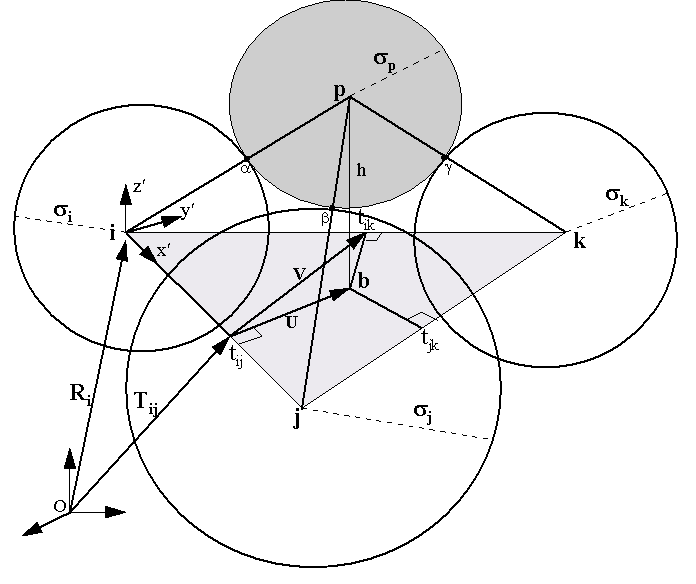
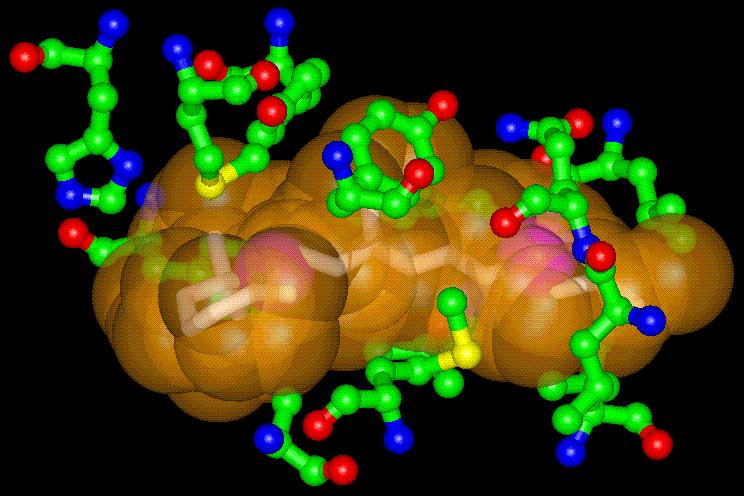
PASS (Putative Active Sites with Spheres) is a simple computational tool
that uses geometry to characterize regions of buried volume in proteins
and to identify positions likely to represent binding sites based upon
the size, shape, and burial extent of these volumes. PASS'S utility as
a predictive tool for binding site identification is tested by predicting
known binding sites of proteins in the PDB using both complexed macromolecules
and their corresponding apo-protein structures. The results indicate that
PASS can serve as a front-end to fast docking. The main utility of PASS
lies in the fact that it can analyze a moderate-size protein (~ 30 kD)
in under twenty seconds, which makes it suitable for interactive molecular
modeling, protein database analysis, and aggressive virtual screening efforts.
As a modeling tool, PASS (i) rapidly identifies favorable regions of the
protein surface, (ii) simplifies visualization of residues modulating binding
in these regions, and (iii) provides a means of directly visualizing buried
volume, which is often inferred indirectly from curvature in a surface
representation. PASS produces output in the form of standard PDB files,
which are suitable for any modeling package, and provides script files
to simplify visualization in Cerius2®, InsightII®,
MOE®, Quanta®, RasMol®, and
Sybyl®. PASS is freely available to all.

Full Text Article
"Fast Prediction and Visualization of Protein Binding Pockets With PASS"
G. Patrick Brady, Jr. and Pieter F.W. Stouten
Journal of Computer-Aided Molecular Design, 14: 383-401, 2000
PDF (Adobe Acrobat)
HTML
Downloads
PASS is free to all in executable form (no guarantees are made or implied, however,
by the author or by DuPont Pharmaceuticals regarding its implementation
or performance).
SGI Irix 6.5
pass10_sgi.tar.gz (gzipped)
pass10_sgi.tar.Z (compressed)
pass10_sgi_n32mips3.tar.gz (gzipped)
pass10_sgi_n32mips3.tar.Z (compressed)
SunOS 5.6
pass10_sun.tar.gz (gzipped)
pass10_sun.tar.Z (compressed)
Linux (2.0.36 and )
pass10_2.0.36linux.tar.gz (gzipped)
pass10_2.0.36linux.tar.Z (compressed)
Running PASS
PASS is trivial to run from the unix command line. To run PASS
on a protein in PDB file "myprotein.pdb," simply type "pass myprotein.pdb."
By default, PASS removes water molecules from the PDB file and removes
all protein hydrogen atoms if less than 20% of the input protein atoms
are hydrogen. PASS offers a host of other options that are available
via command-line flags. A full description
of PASS' usage and command-line flags can be obtained by running "pass"
with no command-line arguments.
Troubleshooting
The most prevalent cause of PASS crashes is nonstandard PDB entries.
For instance, in regions of ambivelent electron density, multiple copies
of certain groups or residues often appear in the PDB file (with a letter
preceding each residue name to distinguish them). E.g. the following snippet
from PDB entry 1hyt (thermolysin):
--------------------------------------------------------------------------------
...
ATOM 58 N GLY 10 21.439 51.310 15.580 1.00 13.52 1HYT 217
ATOM 59 CA GLY 10 21.833 49.927 15.667 1.00 14.69 1HYT 218
ATOM 60 C GLY 10 23.318 49.768 15.939 1.00 22.26 1HYT 219
ATOM 61 O GLY 10 24.055 50.724 16.134 1.00 20.70 1HYT 220
ATOM 62 N ARG 11 23.750 48.533 15.811 1.00 11.91 1HYT 221
ATOM 63 CA ARG 11 25.120 48.151 16.033 1.00 12.60 1HYT 222
ATOM 64 C ARG 11 25.627 47.473 14.816 1.00 16.25 1HYT 223
ATOM 65 O ARG 11 24.959 46.582 14.318 1.00 15.58 1HYT 224
ATOM 66 CB ARG 11 25.156 47.133 17.156 1.00 17.45 1HYT 225
ATOM 67 CG ARG 11 26.230 47.384 18.179 1.00 31.50 1HYT 226
ATOM 68 CD BARG 11 26.273 46.349 19.286 0.50100.00 1HYT 227
ATOM 69 CD AARG 11 26.191 46.343 19.322 0.50 5.13 1HYT 228
ATOM 70 NE BARG 11 25.496 46.791 20.423 0.50 30.12 1HYT 229
ATOM 71 NE AARG 11 25.291 46.713 20.425 0.50 38.48 1HYT 230
ATOM 72 CZ BARG 11 25.769 47.887 21.131 0.50 36.09 1HYT 231
ATOM 73 CZ AARG 11 24.439 45.887 21.057 0.50 21.98 1HYT 232
ATOM 74 NH1BARG 11 26.855 48.650 20.886 0.50 42.74 1HYT 233
ATOM 75 NH1AARG 11 24.327 44.602 20.731 0.50 24.67 1HYT 234
ATOM 76 NH2BARG 11 24.925 48.213 22.113 0.50 16.46 1HYT 235
ATOM 77 NH2AARG 11 23.661 46.370 22.045 0.50 14.65 1HYT 236
ATOM 78 N GLY 12 26.831 47.801 14.410 1.00 11.29 1HYT 237
ATOM 79 CA GLY 12 27.396 47.171 13.215 1.00 11.71 1HYT 238
ATOM 80 C GLY 12 28.206 45.906 13.524 1.00 11.99 1HYT 239
ATOM 81 O GLY 12 28.298 45.433 14.682 1.00 14.99 1HYT 240
...
--------------------------------------------------------------------------------
To remedy such problems, edit the PDB file so as to force it to be standard.
In this case, delete either the "A" or "B" copy of ARG 11 and blank-out the
letter preceding the residue name.
Please email me bugs and/or troubleshooting tips so that I can rectify the
code or include an appropriate suggestion on this page. Thanks.
Financial support for this project was provided by DuPont Pharmaceuticals Co.
 G.Patrick.Brady@dupontpharma.com G.Patrick.Brady@dupontpharma.com
|


{kind=link}
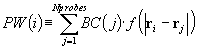{kind=link}
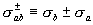{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}