
| http://www.ccl.net/cca/documents/molecular-modeling/node10.shtml |

|
CCL node10 |
|
Next: References Up: Molecular Modeling Previous: Molecular Dynamics and Monte
Molecular ComparisonsDrug design in most cases involves analysis of a series of active and inactive molecules in the search for their mode of action. Since the chemical identity and structure of the receptor protein is frequently unknown, the only clues as to the mechanism of drug action are provided by the drug molecules themselves. In the first step, the molecules have to be superimposed. What seems to be a straightforward task is one of the most difficult and subjective operations. Before one attempts to superimpose molecules, one must choose the criteria for aligning the molecules. This implies at least some working hypothesis on their mode of action, the very reason for aligning the molecules. This is why, alignment rules are constantly revised in the course of studying the problem until consistency between the alignment rules and the hypotheses of drug action is achieved. The major additional difficulty is the flexibility of molecules. At room temperature, molecules containing chains of single bonds exist as a mixture of a large number of conformers of very similar energies. Torsional angles around single bonds are usually very soft and span large ranges of values due to thermal motions. Moreover, interactions with other molecules (e.g., solvent, macromolecules, ions, etc.) can influence the conformation even further. For these reasons, rigid analogs bring the most information in the early stage of analysis.
There are several approaches to fitting molecules. The fit usually involves superimposing chosen atoms of one molecule onto corresponding atoms of the other molecule. The atoms (or dummy atoms representing some groups) are those assumed to be the primary targets for interaction with the corresponding atoms (or groups) in the macromolecular receptor. The simplest approach is to fit two molecules, A and B, as if they were rigid entities. The fitting program will request a list of pairs of atoms to be superimposed. Usually one molecule is treated as a reference (i.e., it is kept immobile) and the second molecule is reoriented in such a way that the sum of the squared distances between atom pairs is at its minimum. Obviously, you need at least three atom pairs for the fit to be possible. Sometimes the option is provided to assign different statistical weights to atom pairs to make the fit tighter for some atoms and more relaxed for others:
where
where components of the translation vector
where The rigid fit of molecules has only limited value for flexible molecules, since the purpose of performing the fit is to examine the possibility of two or more molecules exhibiting the same spatial arrangement of chosen groups. Even a small change in the torsional angle (which is in most cases very inexpensive in terms of the potential energy of the molecule) can dramatically improve the quality of fit. An approach in which the chosen torsional angles are allowed to vary to enhance the fit was reported by Barino (1981). It is an efficient and fast method but suffers occasionally from the fact that the torsional angles are changed solely on the criterion of satisfying the fit, and the potential energy of the modified molecule is not evaluated. Since unrealistic conformations, with bad van der Waals contacts, may sometimes be created, the results of the fit should be carefully examined (e.g., by molecular mechanics).
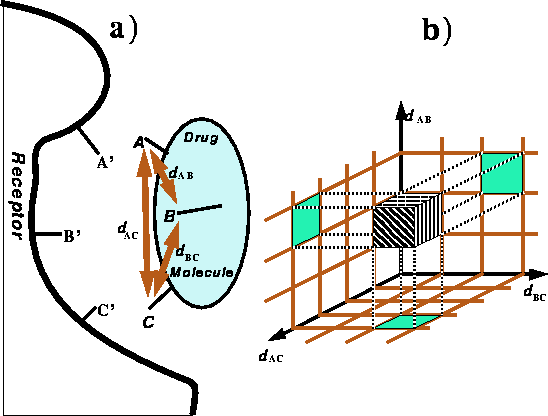
Molecular fit with allowance for adjustment of torsional angles is the core of an ``active analog approach'' in which bad van der Waals contacts are examined (Marshall et al., 1979). For a more formal description of topological and algorithmic issues involved in this approach the reader is refered to Motoc et al. (1986). The method is aimed at cases in which the chemical identity and geometry of the receptor is unknown, i.e., the most common situation in practical drug design. It is based on the concept of the pharmacophore, i.e., the three dimensional arrangement of functional groups essential for recognition and/or activation. The concept of the pharmacophore is illustrated in Fig. 6.29a for the simplest case of three pharmacophoric groups. In this approach, it is assumed that a series of drug molecules (or enzymatic substrates), acting on the same receptor by the same mode of action should present their pharmacophoric groups in a similar three dimensional arrangement for the recognition to take place. The initial step of applying this approach is to identify possible pharmacophoric groups. The next logical step is to check if there is a three dimensional arrangement of these groups common to all active molecules in the series. The three dimensional arrangement of the groups is specified here as a set of distances between the groups. While the first step is usually based on chemical intuition and indirect information about the biological and chemical nature of the receptor, the next step is purely computational and requires a systematic conformational search to be performed on all molecules in the series. The systematic conformational search procedure involves a stepwise change in all designated torsional angles in the molecule by a small increment. The torsional angles allowed to vary correspond to rotatable bonds, i.e., single bonds with low torsional energy barriers (see Fig. 6.21). This systematic search is sometimes called a rigid geometry search in order to emphasize the fact that only torsion angles are allowed to vary, while bond lengths and valence angles are kept fixed at their original values. In this method only those conformations are recorded which do not result in bad van der Waals contacts between non-bonded atoms. Another version of this method called systematic energy search also exists. In the energy search variant, the rigid search of torsional angles is performed and only those conformations whose potential energies are within the chosen energy threshold from the initial conformation are recorded. In another variant of energy search, the zero energy corresponds to the latest lowest energy found during the search. Needless to say, the energy search is much more computationally expensive than the bump checking method since computing all non-bonded and torsional terms in potential energy requires many more operations than checking interatomic distances affected by torsional angles around rotatable bonds. Formally, the bonds designated for torsional scanning in the systematic search procedure cannot be part of a ring, or in topological language, the graph formed from the designated rotational bonds must represent a tree. Changing the torsional angle for a bond which is a member of a ring will invariably lead to changing bond lengths and bond angles. For this purpose, the existing search programs carefully check the topology of the molecule and the relation of rotational bonds. However, there is a method which allows searching for conformations of rings by formally opening a ring and inserting a distance constraint equal to the length of the removed bond. In practice, the distance constraint for the ring search has some tolerance range (e.g. 0.1 Å) since otherwise it is unlikely that the constraint would be satisfied for any but original values of the torsional angles.
To ensure that the whole torsional
space available to the molecule is adequatly sampled, the increment for
changing torsional angles should not be too large, and a value of
5
For example, for
5 torsional angles and There are efficient algorithms rooted in distance geometry which can substantially decrease the number of generated conformations by a priori solving the appropriate trigonometric inequalities and rejecting ranges of torsional angles which would lead to unacceptable conformations. However, even with the most efficient algorithms the computational effort is still very substantial for large molecules with many flexible bonds. Dramatic improvements can be achieved however, by employing distance maps within the ``active analog approach'' as described below.
The set of all valid conformations generated by a systematic search procedure
for a larger molecule has a limited value.
What can you do with a few billion conformations?
The ``active analog approach'' provides a tool for effective analysis
of this plethora of conformations. To start, we need a ``pharmacophore
hypothesis'', i.e., the pharmacophoric groups in each molecule have to
be designated. These groups are chosen as candidates for interaction
with groups on the receptor.
According to the ``active analog approach'', there should
be at least one three dimensional arrangement of these groups common to all
molecules in the series, otherwise the assumed mechanism of recognition
would not be valid.
This arrangement is represented as a set of pairwise
distances between these groups.
The conformational
search is conducted for molecule 1 in the series, and
for each valid conformation, the distances between all specified
pharmacophoric groups are saved together with the set of torsional angles
for this conformation. In the case of 3 groups there are
3 distances (i.e., a triangle) which specify the mutual orientation
of these groups. For 4 groups, there are 6 distances needed to specify
three dimensional orientation of groups. In general, 3M-6 distances are
needed to uniquely specify mutual three dimensional orientation of
M groups
In actual calculations, the distances are represented as a grid
of values, i.e., a distance map (see Fig. 6.29b). For the case
of three pharmacophoric groups
the distance map can be
represented as a three dimensional parllelepiped divided
into bricks. For more than three 3 pharmacophoric groups, the map has to be
built in a space of more than 3 dimensions and
cannot be shown graphically on the computer screen in its
entirety. Only projections containing distance maps of up to three
distances can be displayed. Currently used algorithms allow more than
ten distances to be combined in a distance map.
The edges of the parallepiped circumscribing the distance
map correspond to the allowed ranges for the distances between
pharmacophoric groups, i.e., start at some minimum distance, The purpose of creating a grid is to convert the set of distances into a brick number which is much easier to operate on and compare. Note, that due to small differences between bond lengths and valence angles, there is practically no chance that any two distances between corresponding groups in different molecules will be exactly equal to each another. They may be very close, but most likely never identical. On the other hand, we are not seeking identical distances but similar distances. Identifying a set of distances with a particular brick it an easy and efficient way to classify two sets of distances as either equivalent or different. The assignment of a brick in a distance map to a set of distances is shown in Fig. 6.29b. The search for a common set of distances can be speeded even further if we are not interested in all conformations which realize a given set of distances. When a distance map is used as a constraint, each nonempty brick from the map is translated into several sets of torsional angles. Depending on the topology of the molecule, the size of the brick in the distance map, and the increment used to scan the angles, the single brick may correspond to many different conformations of the molecule. Only some of them will be valid, i.e., do not result in bad van der Waals ``bumps''. In this method we may abondon exploration of the next set of torsional angles for a current brick after the first valid conformation was found. In the case of the first molecule in the series, we can prune the search at the first valid conformation for a given brick, and move to check if there are valid conformations corresponding to the next empty brick in the distance map. After running the search in this way for all the molecules in the series we can rerun the search using a final distance map as a constraint, but this time collect all valid conformations for future analysis. There are basically three possible results from running the systematic search within the active analog approach:
The systematic search is a powerful approach for studying relations between molecules. Since the search explores all possible conformations, it is in principle capable of finding all minima on the conformational energy surface. In most cases, the conformations resulting from a systematic search are used as a starting point for further processing with molecular mechanics, molecular dynamics or quantum approaches to relax the possible strain brought by the rigid geometry regime of the search. The added advantage of the systematic search in its van der Waals bumps checking version, is the small number of actual parameters needed for the run (only van der Waals radii are needed) as compared with molecular mechanics or dynamics which require many constants in the potential energy function. You should not however oversee the limitations of the method related to its computational intractability for larger molecules and the rigid geometry approach. Due to computational expense, the actual sampling increments are usually too large to claim that all possible conformations were explored. The rigid geometry approach prevents the method from finding conformations which could only be realized by changes in bond lengths and valence angles.
The systematic search is not the only method of exploring conformational
space of the molecule, however it is the only method of doing
it systematically,
i.e., checking, in principle, all possible conformations. There are other
approaches which will yield either exactly one,
or more (but not necessarily all) conformations satisfying some requirements.
The distance geometry method (see e.g., Crippen and Havel, 1988;
and Havel et al., 1983) will yield any number of conformations
which satisfy a set of distance constraints.
The distance constraints can
be specified either as discrete values (e.g., bond lengths, 1-3 atom distances)
or ranges. The ranges are usually given for atoms separated by more than
2 bonds (i.e., non-bonded atoms) though discrete values can also be used
for these atoms if we are interested in a particular arrangement of groups
or if we want to keep them fixed (as in ring systems or double and
triple bonds).
For atoms separated by more than 3 bonds, the lower bound of a distance
range is usually taken as the sum of their respective van der Waals radii.
The upper bounds should be set to the distance of the fully extended
conformation for sampling efficiency. The distance constraints are usually
given as an
1 2 3 4 5 6
1 0.0 | 1.5 2.5 3.8 2.5 1.5 C1
____|_____ / \
2 1.5 | 0.0 | 1.5 2.5 3.8 2.5 / \
|_____|_____ / \
3 2.5 1.5 | 0.0 | 1.5 2.5 3.8 C2 C6
|_____|_____ | |
4 2.6 2.5 1.5 | 0.0 | 1.5 2.5 | |
|_____|_____ | |
5 2.5 2.6 2.5 1.5 | 0.0 | 1.5 C3 C5
|_____|_____ \ /
6 1.5 2.5 2.6 2.5 1.5 | 0.0 \ /
| \ /
C4
Figure 6.30: An example of distance constraints for distance geometry
for carbon skeleton for cyclohexane molecule. The upper triangle
and lower triangles contain upper and lower bounds on distances,
respectively.
By setting the upper and lower bound for a distance equal to each other
we efectively freeze this distance. Note however, that the matrix in
Fig. 6.30 contains bounds for
The simple mathematical form of the empirical potential energy function
invites augmenting the ``real'' energy terms for bonded and
non-bonded interactions with the artificial terms which can guide
molecular mechanics optimization or molecular dynamics trajectories towards
some specific conformation. These artifical terms are called constraints
or restraints,
and they may have many uses. The simplest application of constraints in
molecular mechanics is to enforce a value of some geometric parameter
(like interatomic distance or angle) on the final structure. The
constraint,
where
When the potential energy function with constraining terms is submitted to minimization, the compromise between constraints and ``real'' potential energy terms is found. If enforcing the constraint will only require changing ``soft'' geometrical parameters (e.g., if the torsional angle change can satisfy the constraint) the energy, and also bond lengths and valence angles, resulting from constrained minimization will be close to the ones from unconstrained minimization. However, in some cases adding constraints might result in an unrealistically distorted molecule. The easiest way to check this is to submit the geometry resulting from the constrained run to geometry optimization but without constraints. If the difference between two energies is small (e.g., less than 5 kcal/mol) we can asume that the constraint can be satisfied. A large energy difference suggests that the constraint introduces a substantial strain. In this case we should carefully examine the starting geometry to asses if there is in fact any chemically meaningful way in which the constraint can be satisfied. If the molecule can exists in several stable conformations separated by high energy barriers (e.g., cis/trans along the double bond), the starting conformation should be the one which has a chance to satisfy the constraint. The mathematical form of constraints introduced into the potential energy function depends upon the problem. The simple harmonic constrains given by eq. 6.64 are the most popular but not always best. For example, the NOE constraints derived from two-dimensional NMR represent frequently an averaged distance between related atoms (e.g. hydrogens of freely rotating methyl group). Moreover, the magnitude of the Nuclear Overhauser Effect is proportional to the inverse of the sixth power of the distance between atoms. That is why, the NOE will yield much more accurate results for smaller distances and only rough estimates for larger ones. For this reason, the appropriate constraint should penalize the energy more on the small distance side and less for the larger distances. Clearly, a simple parabola centered around the NOE distance is not appropriate here, and some asymmetrical function is needed. Some constraints are also given as ranges, e.g., of atom distances. The simple approach, which would add the penalty to energy for distances outside the given range and not add it within a range, is inappropriate since it produces a discontinuity in energy function at the range limits. The energy function has to be continuous function of coordinates if the derivative based minimization techniques are used. Sometimes a trick is used in which this rectangular shape function is still used, but the derivatives of the constraining term are not calculated. This will affect, however, more complex (and more efficient) minimization algorithms and can even cause their divergence. The constraints are therefore frequently constructed by pasting together a constant function with a polynomial which in the vicinity of a range limit smoothly rises the energy from zero to some positive constant. The constraints can also be used in molecular mechanics to superimpose molecules. There are basically two posibilities. The simpler of the two is when one of the molecules (say A) is chosen as a template (sometimes called a reference molecule) and the other (say B) is optimized with the constraint that the selected atoms are forced to occupy the positions of the corresponding atoms of the template. In this case simple harmonic constraints are used:
where
The other possible approach is to look for the best fit of atoms in the series of molecules. In this case there is no reference molecule. One such approach was developed by Labanowski et al. (1986). The outline of this approach is given in Fig. 6.31. In this case, the atoms are fitted not to each another but to geometrical targets called reference points. The reference point is basically an average of atom positions belonging to the set of atoms to be superimposed. In the example in Fig. 6.31 there are three molecules: A, B, and C and four sets of superimposed atoms: 1, 2, 3, and 4. The reference points are denoted here by R. For each molecule in the series, the geometry is optimized with a potential energy function augmented with the following artificial terms shown here for a molecule A:
where Before this approach can be used, the initial reference point positions must be found. This is done by performing a rigid fit of all molecules to the first molecule in the series and finding the average position of atoms in each set of superimposed atoms (including atoms of the first molecule). For this reason, the most rigid molecule should be chosen as the first in the series. The energy of each molecule is then minimized and reference point positions incremented by the portion of the new atom coordinates. Assuming that a molecule M has just been optimized, the reference points are updated according to the following formula:
where
where the sum runs over optimized molecules in the first cycle of
optimization, and over all molecules in the second and all subsequent cycles.
To guard against drifting of the whole molecular assemblage, one of the
reference points is kept fixed, namely the one for which the sum
In conclusion, there are many approaches to superimposing molecules. Though we would all prefer the single approach which works in all situations, it should now be obvious to the reader that such an ideal method will probably never exist. Each superimposing problem is different and hence, for each problem, the best method should be used. The above methods can also be used in the case when the molecular structure of the receptor is known. In this case we can introduce a degree of flexibility also into the receptor site, probing the various ways in which the ligand can be ``docked'' into the receptor. It is also evident from the above discussion that the fully automatic method of mapping receptor sites is still behind the horizon. The successful computer-aided drug design will for a forseeable future require an experienced and insightful chemist and a lot of auxiliary experimental information.
Next: References Up: Molecular Modeling Previous: Molecular Dynamics and Monte Computational Chemistry Wed Dec 4 17:47:07 EST 1996 |
[ CCL Home Page ]
[ About CCL ]
[ Resources ]
[ Search CCL ]
[ Announcements ]
[ Links ]
[ E-mail us ]
[ Raw Version of this page ]
| Modified: Sat May 23 16:00:00 1998 GMT |
| Page accessed 10111 times since Sat Apr 17 12:48:52 1999 GMT |
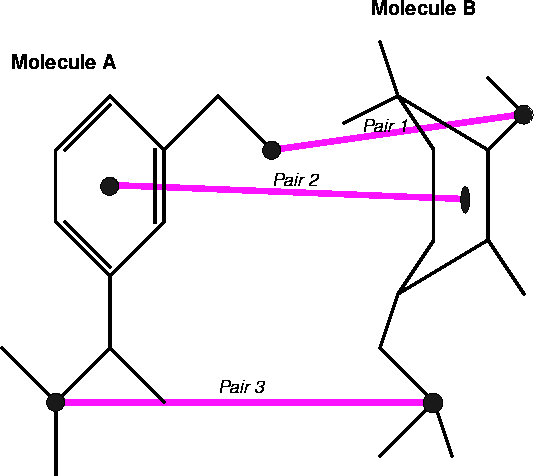
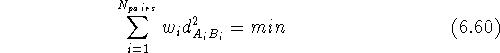
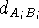 is the distance between the atoms of molecule A and B,
respectively,
and
is the distance between the atoms of molecule A and B,
respectively,
and 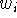 is the statistical weight of the i-th atom pair. Mathematically,
the problem of the rigid fit of two molecules can be solved in a number of
ways varying in their robustness. As is evident from
eq.
is the statistical weight of the i-th atom pair. Mathematically,
the problem of the rigid fit of two molecules can be solved in a number of
ways varying in their robustness. As is evident from
eq. 
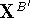 , of molecule B
are then calculated from the original coordinates,
, of molecule B
are then calculated from the original coordinates, 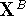 as:
as:
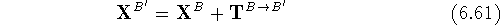
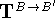 are
added to corresponding coordinates for all atoms of molecule B.
The next step involves stepwise construction of the rotation matrix,
are
added to corresponding coordinates for all atoms of molecule B.
The next step involves stepwise construction of the rotation matrix,
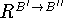 , which superimposes the translated coordinates
, which superimposes the translated coordinates
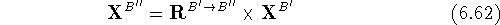
 are the translated and final
(i.e., transformed) atomic coordinates of molecule B''.
More elegant and noniterative methods are based on linear
algebra and singular value decomposition of the product
of matrices of cartesian
coordinates of two molecules whose geometric centers were superimposed.
These methods however, can sometimes invert the molecule
and change its chirality. Probably the most efficient method
for the rigid fit of molecules,
though at the same time the least popular, is based on the concept of
quaternions (see e.g., Altman, 1986).
It is currently the fastest, noniterative method which will
not change the chirality of the fitted molecule.
are the translated and final
(i.e., transformed) atomic coordinates of molecule B''.
More elegant and noniterative methods are based on linear
algebra and singular value decomposition of the product
of matrices of cartesian
coordinates of two molecules whose geometric centers were superimposed.
These methods however, can sometimes invert the molecule
and change its chirality. Probably the most efficient method
for the rigid fit of molecules,
though at the same time the least popular, is based on the concept of
quaternions (see e.g., Altman, 1986).
It is currently the fastest, noniterative method which will
not change the chirality of the fitted molecule.
 is considered an upper limit for meaningful computations.
Using larger increments brings the danger of skipping over valid
conformations which may exist for intermediate values of torsional
angles. This danger cannot be avoided even for very fine
is considered an upper limit for meaningful computations.
Using larger increments brings the danger of skipping over valid
conformations which may exist for intermediate values of torsional
angles. This danger cannot be avoided even for very fine 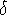 increments,
since some compact, yet energetically preferred conformations which are
found by
explicit geometry optimization of all degrees of freedom, are realized by small
changes in valence angles and bond lengths allowing non-bonded atoms to
avoid close van der Waals contacts. Moreover, molecules (especially
the larger ones) have a natural tendency
to form compact conformations since they result in the largest overall value
of London dispersion forces (see Fig.
increments,
since some compact, yet energetically preferred conformations which are
found by
explicit geometry optimization of all degrees of freedom, are realized by small
changes in valence angles and bond lengths allowing non-bonded atoms to
avoid close van der Waals contacts. Moreover, molecules (especially
the larger ones) have a natural tendency
to form compact conformations since they result in the largest overall value
of London dispersion forces (see Fig. 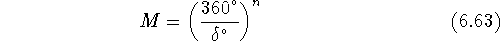
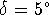 the number of generated
conformations is
the number of generated
conformations is 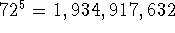 . For each conformation all
pairwise distances between non-bonded atoms are checked against the
sum of van der Waals radii of the two atoms
and the conformation is accepted if
none of the distances is smaller than this value. The allowed conformations
usually form families, i.e., they come in groups representing contiguous
ranges of torsional angles. On an angle map
. For each conformation all
pairwise distances between non-bonded atoms are checked against the
sum of van der Waals radii of the two atoms
and the conformation is accepted if
none of the distances is smaller than this value. The allowed conformations
usually form families, i.e., they come in groups representing contiguous
ranges of torsional angles. On an angle map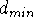 , and
end at some maximum distance,
, and
end at some maximum distance, 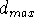 . For real atoms, the distance
. For real atoms, the distance 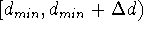 ,
, 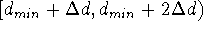 ,
...
,
... 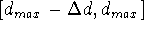 . By dividing edges of the parallelepiped
circumscribing the distance map we effectively divide the map into a series
of elementary bricks. The number of subsections should be carefully chosen.
Choosing this value too large will result in the loss of information.
Making it too small will produce artifacts described below.
The size of
. By dividing edges of the parallelepiped
circumscribing the distance map we effectively divide the map into a series
of elementary bricks. The number of subsections should be carefully chosen.
Choosing this value too large will result in the loss of information.
Making it too small will produce artifacts described below.
The size of 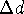 depends upon the increment
depends upon the increment 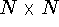 matrix whose one triangle consists of the upper bounds
for distances and the other for lower bounds, while the diagonal is
filled with zeros. Frequently hydrogen atoms are omitted to speed up the
search for valid conformations. For an example of a matrix of distance
constraints refer to Fig.
matrix whose one triangle consists of the upper bounds
for distances and the other for lower bounds, while the diagonal is
filled with zeros. Frequently hydrogen atoms are omitted to speed up the
search for valid conformations. For an example of a matrix of distance
constraints refer to Fig. 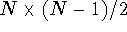 distances.
For molecules larger than 4 atoms
distances.
For molecules larger than 4 atoms 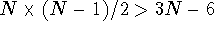 , that is,
there are more distances than internal coordinates. In effect, the system
is overdetermined if all of the distances were fixed. In such a case the
distance geometry would not find an acceptable conformation if the
distances were not consistent with each other. Using ranges for some
distances instead
of exact values produces an infinite number of possible conformations, unless
some distancess violate the triangle inequality:
, that is,
there are more distances than internal coordinates. In effect, the system
is overdetermined if all of the distances were fixed. In such a case the
distance geometry would not find an acceptable conformation if the
distances were not consistent with each other. Using ranges for some
distances instead
of exact values produces an infinite number of possible conformations, unless
some distancess violate the triangle inequality: 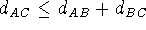 .
For this reason the distance geometry approach is not a systematic approach
since it cannot produce all possible
distances (there is an infinite number of them) and only provides us
with a sample of conformations.
The conformations are in
principle created at random but they satisfy the distance
constraints.
The program runs until a requested number of conformations is generated.
If none of the conformations obtained is of interest you may run program
again and obtain an additional sample of conformations.
The major strength of the distance geometry approach is that it will
always provide a solution if there is one (i.e., when distances are not
conflicting), contrary to the systematic
search which might fail to detect a valid conformation if the sampling
interval
.
For this reason the distance geometry approach is not a systematic approach
since it cannot produce all possible
distances (there is an infinite number of them) and only provides us
with a sample of conformations.
The conformations are in
principle created at random but they satisfy the distance
constraints.
The program runs until a requested number of conformations is generated.
If none of the conformations obtained is of interest you may run program
again and obtain an additional sample of conformations.
The major strength of the distance geometry approach is that it will
always provide a solution if there is one (i.e., when distances are not
conflicting), contrary to the systematic
search which might fail to detect a valid conformation if the sampling
interval  , may be a simple harmonic term:
, may be a simple harmonic term:
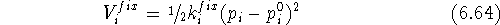
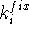 is the force constant whose value represents the strength
with which we want to enforce the constraint,
is the force constant whose value represents the strength
with which we want to enforce the constraint, 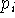 denotes the actual
value of geometrical parameter being constrained or enforced and
denotes the actual
value of geometrical parameter being constrained or enforced and 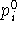 is the target value of the parameter. For practical reasons, two types of
constraints can be distinguished even though their mathematical form
may be identical:
is the target value of the parameter. For practical reasons, two types of
constraints can be distinguished even though their mathematical form
may be identical:
 Å
Å  ) will be appropriate.
) will be appropriate.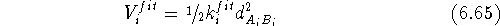
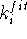 is the force constant whose value represents the weight which
we assign to a quality of the fit for i-th pair, and
is the force constant whose value represents the weight which
we assign to a quality of the fit for i-th pair, and 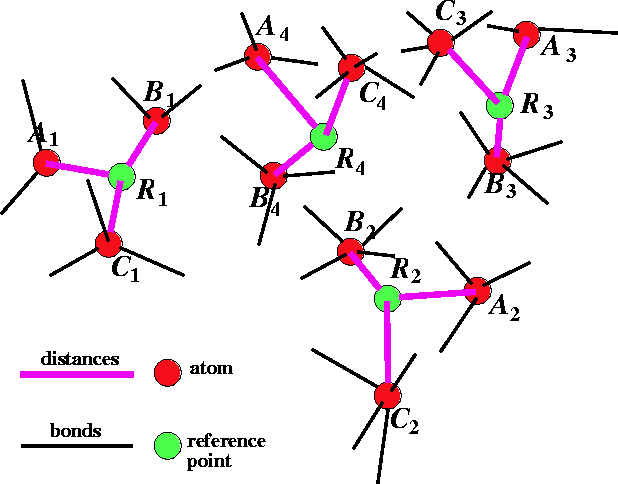
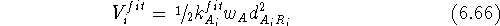
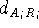 is the distance between atom of the
molecule A belonging to
the i-th set of superimposed atoms, and a reference point
is the distance between atom of the
molecule A belonging to
the i-th set of superimposed atoms, and a reference point 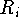 for this set.
The
for this set.
The 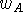 is the statistical weight of the molecule A and
is the statistical weight of the molecule A and 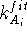 is the force constant for this atom of molecule A. The value of this term
is always positive except when the atom and the reference point are
exactly on top of each another. The magnitude of its contribution
to energy depends upon the
value for the fitting force constant,
is the force constant for this atom of molecule A. The value of this term
is always positive except when the atom and the reference point are
exactly on top of each another. The magnitude of its contribution
to energy depends upon the
value for the fitting force constant,  mol
mol 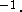 Å
Å 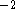 are used but the force constants should be individualized to reflect the
chemical nature of pharmacophoric group.
Again, the force constants are not meant to reflect
how hard it is to change the atom position with respect to the other atoms of
the molecule, since this information is provided directly by the
potential energy function. These values should rather reflect the assumed mode
of interaction of a given atom/group in the molecule with its counterpart
in the receptor and its overal contribution to the total binding energy.
For example,
hydrogen binding requires a precise orientation of groups but on the
other hand contributes only roughly about 5 kcal/mol to the binding energy
(though in solvent free space the contribution may be much higher).
The electrostatic interactions of two ions (e.g. salt bridges) can contribute
substantially to binding energy if the maginitude of net atomic charges
involved is large. On the other hand this interaction does not depend
steeply on the relative orientation of the charges involved.
The magnitude of dipole/dipole
interactions is large for closely spaced dipoles
and also depends strongly on orientation. You can
mimic the orientation of a dipole by fitting both atoms belonging to the
bond providing the dipole and using a larger (e.g. twice as large) fitting
force constants for these atoms. You might use a similar technique for
hydrogen bonds to incorporate orientational information but in this case
you might want to reduce the force constants slightly to reflect the
fact that the two atoms are fitted per each hydrogen bond.
You can also use constructs of dummy atoms and fit them together if the
suspected mode of interaction between molecule and receptor is not of
a simple category.
are used but the force constants should be individualized to reflect the
chemical nature of pharmacophoric group.
Again, the force constants are not meant to reflect
how hard it is to change the atom position with respect to the other atoms of
the molecule, since this information is provided directly by the
potential energy function. These values should rather reflect the assumed mode
of interaction of a given atom/group in the molecule with its counterpart
in the receptor and its overal contribution to the total binding energy.
For example,
hydrogen binding requires a precise orientation of groups but on the
other hand contributes only roughly about 5 kcal/mol to the binding energy
(though in solvent free space the contribution may be much higher).
The electrostatic interactions of two ions (e.g. salt bridges) can contribute
substantially to binding energy if the maginitude of net atomic charges
involved is large. On the other hand this interaction does not depend
steeply on the relative orientation of the charges involved.
The magnitude of dipole/dipole
interactions is large for closely spaced dipoles
and also depends strongly on orientation. You can
mimic the orientation of a dipole by fitting both atoms belonging to the
bond providing the dipole and using a larger (e.g. twice as large) fitting
force constants for these atoms. You might use a similar technique for
hydrogen bonds to incorporate orientational information but in this case
you might want to reduce the force constants slightly to reflect the
fact that the two atoms are fitted per each hydrogen bond.
You can also use constructs of dummy atoms and fit them together if the
suspected mode of interaction between molecule and receptor is not of
a simple category.
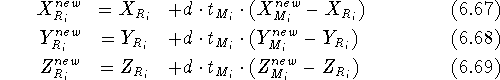
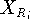 and
and 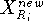 is the previous and updated X- coordinate
of the i-th reference point,
is the previous and updated X- coordinate
of the i-th reference point, 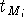 is the scaling factor, d is
the damping factor and
is the scaling factor, d is
the damping factor and 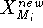 is the new X coordinate
of the atom
is the new X coordinate
of the atom 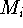 which has just been calculated.
The damping factor,
which has just been calculated.
The damping factor, 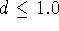 , is usually 1.0 but can
be made smaller if the starting coordinates of atoms being superimposed
are very different from molecule to molecule and the reference point
positions exhibit the oscillatory behaviour. The scaling factor
, is usually 1.0 but can
be made smaller if the starting coordinates of atoms being superimposed
are very different from molecule to molecule and the reference point
positions exhibit the oscillatory behaviour. The scaling factor 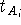 for the i-th atom of some molecule A of the series
represents the weight with which the coordinates of this atom
influence the position of a reference point and is given by the following
expression:
for the i-th atom of some molecule A of the series
represents the weight with which the coordinates of this atom
influence the position of a reference point and is given by the following
expression:
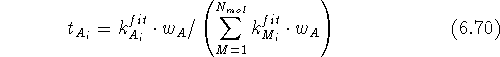
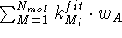 has the maximum value of all
reference points.
The optimization is stopped when the energy of optimized molecules
is no longer changing compared to their energy in the previous cycle.
has the maximum value of all
reference points.
The optimization is stopped when the energy of optimized molecules
is no longer changing compared to their energy in the previous cycle.